Methanogenese
| Übergeordnet |
| Anaerobe Atmung Biosynthese der Alkane Metabolismus des Methan |
| Untergeordnet |
| aus Acetat aus CO2 aus Methanol aus Methylaminen |
| Gene Ontology |
|---|
|
|
Die Methanogenese (auch Methanbildung) ist die Bildung von Methan durch den Stoffwechsel von Lebewesen, die als Methanogene oder Methanbildner bezeichnet werden. Sie findet – bis auf wenige Ausnahmen – größtenteils in der letzten Stufe des anaeroben, mikrobiellen Abbaus von Biomasse statt. Dabei setzen die meisten Methanbildner Kohlenstoffdioxid und Wasserstoff zu Methan um. Auch aus einfachen organischen C1-Verbindungen wie Ameisensäure, Methanol und Methylaminen wird Methan gebildet. Essigsäure wird durch Essigsäure-spaltende (acetoklastische) Methanbildner in Methan und Kohlenstoffdioxid umgewandelt. Andere bakterielle Gärungsprodukte wie Milchsäure, Propionsäure und Buttersäure können dagegen nicht als Ausgangsstoffe für die Methanbildung verwendet werden.
In der Literatur wird die Methanogenese überwiegend als spezifischer, anaerober Stoffwechselweg von Archaeen behandelt, als eine spezielle Form der anaeroben Atmung. Diese nutzen dabei die exergone (energiefreisetzende) Methanogenese als Energiequelle.[1] Daher fokussiert der Artikel die anaerobe Methanfreisetzung in Archaeen.
Bedeutung
Die Methanogenese ist ein zentraler Bestandteil des Kohlenstoffzyklus der Erde, da die entstehenden Abbauprodukte, Methan und ggf. Kohlenstoffdioxid, wieder in diesen Kreislauf gelangen. Da diese Gase, insbesondere Methan, wirksame Treibhausgase sind, hat die Methanogenese auch bei der Vermeidung der globalen Erwärmung an Bedeutung gewonnen.[2] Vermutlich spielt die biotische Methanproduktion auch eine Rolle bei der Entstehung von Methanhydrat, dessen wirtschaftliche Nutzung von Interesse ist.
Eine wichtige Bedeutung hat die Methanogenese im Ablauf und Ende der anaeroben Nahrungskette, da sie das Wachstum vieler syntropher Bakterien überhaupt erst ermöglicht. Diese sekundären Gärer gewinnen ihre Energie aus der Umsetzung von Lactat, Propionat, Butyrat und einfachen organischen Verbindungen, wobei neben Kohlenstoffdioxid und Acetat auch Wasserstoff entsteht. Aus thermodynamischen Gründen sind jedoch diese Vergärungsreaktionen nur möglich, wenn der dabei entstehende Wasserstoff rasch wieder verbraucht wird und der H2-Partialdruck nicht über 100 Pa ansteigt. Das wird durch in enger Nachbarschaft lebende Methanogene gewährleistet, die diesen Wasserstoff wiederum für die Methanogenese benötigen. Der Transfer von Wasserstoff zwischen syntrophen Bakterien und den Archaeen, also zwischen verschiedenen Spezies, bezeichnet man auch als Interspezies-Wasserstofftransfer.[3][4]
Da Methanogene, vergesellschaftet mit syntrophen Bakterien, auch im menschlichen Verdauungstrakt vorkommen, hat dort die Methanogenese einen Einfluss auf die Verdauung.[5] Etwa 10 % der Anaerobier im Darm des Menschen sind Methanogene der Arten Methanobrevibacter smithii und Methanosphaera stadtmanae. Diese nutzen die beiden Produkte bakterieller Gärungen Wasserstoff und Formiat für die Methanogenese. Eine hohe Konzentration an Wasserstoff hemmt die ATP-Erzeugung anderer Bakterien. M. smithii baut unter Methanbildung auch Methanol ab, das für den Menschen toxisch ist. Daher haben die Methanogenen einen positiven Einfluss auf die menschliche Darmflora. Ob diese auch beeinflussen, wie viel Energie der Mensch aus der Nahrung aufnehmen kann, ist noch Gegenstand der Forschung.
Vorkommen


Die Methanbildung kommt in der Natur in überwiegend anaeroben Milieus vor, in denen ein Abbau von Biomasse stattfindet. Das können beispielsweise Sedimente von Seen und des Meeres, der Pansen von Rindern, der Darm von Termiten und Menschen, Reisfelder oder Sümpfe sein. Sie sind ferner mit Bakterien vergesellschaftet, um deren Stoffwechselprodukte zu nutzen, wie beim Abbau von nassem Holz bei Clostridium butyricum.[6] Auch Kläranlagenschlammbecken als künstliche Anlagen für den biologischen Abbau sind mögliche Orte für eine Methanogenese. In diesen Habitaten herrschen moderate, für mesophile Organismen geeignete Temperaturen. Methanogene wurden auch in Böden entdeckt, die Sauerstoff enthalten; sie können mit dem damit verbundenen oxidativen Stress umgehen.[7] Die Methanogenese tritt auch in Umgebungen mit extrem hohen und niedrigen[8] Temperaturen (z.B. Methanogenium frigidum)[7] sowie bei hohen Salz- oder Säuregehalten auf, beispielsweise in geothermalen Systemen.[4] In allen Fällen müssen in diesen Habitaten die Konzentrationen von Sulfat, Nitrat, Mangan(IV)- und Eisen(III)-Ionen niedrig sein, da anderenfalls Bakterien diese Stoffe als externe Elektronenakzeptoren in einer anaeroben Atmung verwenden und in dieser Atmung die für Methanogene nutzbaren Elektronendonatoren verbrauchen. Die Redoxvorgänge mit diesen Elektronenakzeptoren laufen nämlich bevorzugt vor der Methanogenese ab und den Methanogenen wird dadurch ihre Energiequelle und damit ihre Lebensgrundlage entzogen.[9] Die Methanproduktion in Meeren ist daher vergleichsweise gering, da die in den Meeren gelösten Sulfate in den Sedimenten von sulfatreduzierenden Bakterien unter Verbrauch von Wasserstoff zu Schwefelwasserstoff umgesetzt werden (Desulfurikation).[10]
Unter anaeroben Bedingungen ist Kohlenstoffdioxid, das Substrat der meisten Methanogenen, selten limitierend, da es fortlaufend durch Vergärungsreaktionen durch vergesellschaftete Bakterien freigesetzt wird.[4] Die meisten Methanogenen bevorzugen einen neutralen pH-Wert, Ausnahmen sind beispielsweise Methanocalculus alkaliphilus und Methanosalsum natronophilum, die im Basischen bei einem pH-Wert von 9,5 optimal wachsen sowie Methanoregula booneii, dessen pH-Optimum im Sauren bei 5,1 liegt.[6] Methanosalsum natronophilum toleriert bei gleichhohem pH-Wert einen höheren Salzgehalt als Methanocalculus alkaliphilus.[6]
Klassifizierung
Die Methanogenese wird von Archaeen betrieben, die alle zur Abteilung der Euryarchaeota zählen. Dort werden sie in den Klassen Methanobacteria, Methanococci, „Methanomicrobia“ und Methanopyri geführt. Methanogene Archaeen finden sich dabei in folgenden sechs Ordnungen: Methanopyrales, Methanobacteriales, Methanococcales, Methanomicrobiales, Methanosarcinales und Methanocellales.[9][11][12] Hierbei ist Methanopyrales der stammesgeschichtlich älteste, Methanosarcinales dagegen der phylogenetisch jüngste Zweig. Die 2008 entdeckte sechste Ordnung (Methanocellales) geht auf im Boden von Reisfeldern vorkommenden Archaeen Methanocella paludicola und Methanocella arvoryzae zurück. Diese betreiben eine Methanogenese aus Kohlenstoffdioxid und Wasserstoff. Methanoplasmatales, die mit den Thermoplasmatales verwandt sind, wurden in der Literatur als siebte Ordnung vorgeschlagen,[13] dann aber umbenannt in Methanomassiliicoccales.[6][14]
Methanopyrales, Methanobacteriales sowie Methanococcales zählt man zu den Klasse I-, Methanomicrobiales zu den Klasse II-Methanogenen. Methanosarcinales sind Klasse III-Methanogene.[15]
Substratvielfalt
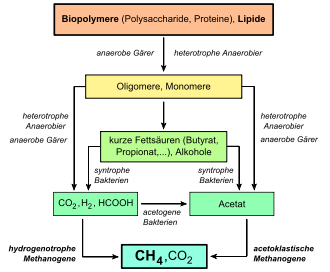
In vielen Habitaten sind Methanogene Endkonsumenten in der sogenannten „anaeroben Nahrungskette“.[16] In dieser Kette werden zunächst Biopolymere wie Proteine und insbesondere Polysaccharide wie Cellulose über Oligomere in Monomere (beispielsweise Aminosäuren und Kohlenhydrate) gespalten. Lipide werden hierbei in ihre Komponenten (beispielsweise Fettsäuren) abgebaut. Danach vergären Bakterien diese Spaltprodukte zu einfachen Carbonsäuren (wie Formiat, Acetat, Propionat, Lactat und Succinat), zu Alkoholen (wie Ethanol, 2-Propanol und Butanol) und zu anderen niedermolekularen Verbindungen (H2, CO2 und kurzkettige Ketone). Syntrophe, acetogene Bakterien nutzen einen Teil dieser Verbindungen und setzen sie zu Acetat und C1-Verbindungen um. Im letzten Teil der anaeroben Nahrungskette, der Methanogenese, werden diese Verbindungen als Kohlenstoff-, Reduktans und Energiequelle verwendet, wobei CH4 und meistens CO2 freigesetzt werden.
- Die meisten Methanogenen betreiben die Methanogenese mit Kohlenstoffdioxid (CO2) als Substrat, bei der Wasserstoff (H2) als primäres Reduktionsmittel verwendet wird.[4] Man bezeichnet solche Methanogene als wasserstoffoxidierend oder hydrogenotroph. Zu den obligaten (ausschließlichen) Hydrogenotrophen zählen die Methanopyrales, Methanobacteriales, Methanococcales und Methanomicrobiales, die nur H2 und CO2 oder Ameisensäure (HCOOH) als Substrate für die Methanogenese nutzen.[17][2] Eine Ausnahme unter den Methanomicrobiales ist Methanosphaera stadtmanae, die im menschlichen Verdauungstrakt vorkommt. Sie ist auf Methanol und Wasserstoff angewiesen, da sie nicht CO2 nutzen kann.[18] Modellorganismen unter den Hydrogenotrophen sind Methanothermobacter thermautotrophicus> und Methanocaldococcus jannaschii (ehemals Methanococcus jannaschii). In Methanomassiliicoccales wurden bisher keine diesbezügliche Methanogenese nachgewiesen.[6] Die hydrogenotrophe Methanogenese tritt besonders in Sedimenten der Tiefsee sowie im Darm von Termiten, Menschen und Tieren auf.[7] Das hierbei entstehende Methan trägt ca. 33 % zur jährlichen Methanproduktion auf der Erde bei.[7]
- Kohlenstoffmonoxid (CO) kann nur von wenigen Arten für die Methanogenese genutzt werden.[4] M. thermoautotrophicus und Methanosarcina barkeri bilden aus vier Molekülen CO drei Moleküle CO2 und ein Molekül Methan. Auch Methanosarcina acetivorans kann CO als Substrat verwenden, wobei parallel Acetat und Formiat gebildet werden.[19] Diese Art der Acetogenese in Methanogenen bezeichnet man als carboxidotrophe Acetogenese.[20]
- Die Methanosarcinales sind die vielseitigsten Methanogenen, sie können sehr unterschiedliche C1-Verbindungen für die Methanogenese verwenden. Neben CO2 + H2 nutzen viele Arten C1-Verbindungen, in denen der Kohlenstoff als Methylgruppe enthalten ist, wie Methanol, Methylamine (Mono-, Di-, Trimethylamin) und Methylthiole (Dimethylsulfid, Methanthiol).[16] Methanosarcinales können aber nicht Ameisensäure umsetzen.
- Acetat (CH3COOH) ist die einzige C2-Verbindung, die für eine Methanogenese genutzt werden kann. Dazu sind – soweit bisher bekannt – nur die Gattungen Methanosaeta und Methanosarcina (Methanosarcinales) fähig. Man bezeichnet sie als acetoklastische Methanogene oder Acetoklaster. Bei dieser Art von Methanogenese wird Acetat in CO2 und CH4 gespalten.[4] Obwohl Acetat nur von wenigen Archaeen für eine Methanogenese genutzt wird, trägt das dabei entstehende Methan mit 66 % zur jährlichen Methanproduktion auf der Erde bei.[21] Damit ist die Methanbildung der Acetoklaster die größte biogene Quelle. Acetoklaster treten vor allem in Faultürmen/Biogasanlagen, Reisfeldern oder Sümpfen auf.[7]
- N-methylierte Amine mit einer C2-Kohlenstoffkette werden von manchen Methanogenen der Gattung Methanococcoides (gehört zu den Methanosarcinales) auch für die Methanogenese verwertet.[22][23] Jedoch werden bei diesen Verbindungen nur die Methylgruppen genutzt. So wird beispielsweise Cholin oder Dimethylaminoethanol (DMAE) zu Ethanolamin abgebaut und die freigewordene Methylgruppe in der Methanogenese verwertet. DMAE wird unter anderem von Methanococcoides methylutens und Methanococcoides burtonii abgebaut. Auch Betain dient manchen Methanococcoides-Arten als Substrat: analog wie beim Cholin wird eine Methylgruppe freigesetzt und zu Dimethylglycin abgebaut. Ob Methanogene auch methylierte Amine mit längeren Seitenketten nutzen können, wird noch untersucht. Die methylotrophe Methanogenese tritt besonders im Meer oder hypersalinen, sulfatreichen Sedimenten auf.[7]
| Reaktion in der Methanogenese | ΔG0’ [kJ/mol CH4][4] | Organismus |
|---|---|---|
| Kohlenstoffdioxid-Typ | ||
| CO2 + 4 H2 → CH4 + 2 H2O | −135 | die meisten Methanogenen |
| 4 HCOOH → CH4 + 3 CO2 + 2 H2O | −130 | viele hydrogenothrophe Methanogene |
| CO2 + 4 C3H8O → CH4 + 4 C3H6O + 2 H2O | −37 | manche hydrogenothrophe Methanogene |
| 4 CO + 2 H2O → CH4 + 3 CO2 | −196 | Methanothermobacter und Methanosarcina |
| mit Methylverbindungen | ||
| 4 CH3OH → 3 CH4 + CO2 + 2 H2O | −105 | Methanosarcina und andere methylothrophe Methanogene |
| CH3OH + H2 → CH4 + H2O | −113 | Methanomicrococcus blatticola und Methanosphaera[24] |
| 2 (CH3)2S + 2 H2O → 3 CH4 + CO2 + 2 H2S | −49 | manche methylothrophe Methanogene |
| 4 CH3NH2 + 2 H2O → 3 CH4 + CO2 + 4 NH3 | −75 | manche methylothrophe Methanogene |
| 2 (CH3)2NH + 2 H2O → 3 CH4 + CO2 + 2 NH3 | −73 | manche methylothrophe Methanogene |
| 4 (CH3)3N + 6 H2O → 9 CH4 + 3 CO2 + 4 NH3 | −74 | manche methylothrophe Methanogene |
| 4 CH3NH3Cl + 2 H2O → 3 CH4 + CO2 + 4 NH4Cl | −74 | manche methylothrophe Methanogene |
| mit Acetat (Essigsäure) | ||
| CH3COOH → CH4 + CO2 | −33 | Methanosarcina und Methanosaeta |
| mit N-methylierten Aminen mit einer C2-Seitenkette | ||
| 4 (CH3)3N+CH2CH2OH + 6 H2O → 4 H2NCH2CH2OH + 9 CH4 + 3 CO2 + 4 H+ | −63[23] | manche Methanosarcina |
| 2 (CH3)2NCH2CH2OH + 2 H2O → 2 H2NCH2CH2OH + 3 CH4 + 3 CO2 | −47[23] | manche Methanosarcina |
| 4 (CH3)3N+CH2COO− + 2 H2O → 4 (CH3)2NH+CH2COO− + 3 CH4 + CO2 | −240[22] | manche Methanosarcina |
Unterscheidung mittels Cytochromen
Methanogene der Ordnungen Methanosarcinales enthalten Cytochrome, während man diese unter den anderen Ordnungen nicht gefunden hat. Dies hat neben physiologische auch stoffwechselspezifische Auswirkungen darauf, wie methanogene Archaeen Kohlenstoffdioxid und Wasserstoff zu Methan verstoffwechseln.[9]
- Methanogene mit Cytochromen enthalten Methanophenazin. Es ist der universelle Elektronenüberträger in der Membran dieser Methanogenen und ersetzt dort Chinon, das nur in geringen Konzentrationen vorkommt und in anderen Organismen für den Transport von Elektronen in der Atmungskette essentiell ist. Viele Methanosarcinales wachsen auf Acetat und methylierten Verbindungen. Falls sie CO2 + H2 verwerten, muss der H2-Partialdruck über 10 Pa liegen. Methanogene mit Cytochromen wachsen langsam, die Teilungsrate liegt bei über 10 Stunden pro Teilung. Bisher wurden unter Methanogenen mit Cytochromen keine Vertreter entdeckt, die unter hyperthermophilen Bedingungen wachsen.
- Bei Methanogenen ohne Cytochromen fehlt dagegen Methanophenazin. Im Gegensatz zu den Methanosarcinales wachsen jene Methanogene mit CO2 + H2 beziehungsweise Ameisensäure und können weder methylierte Verbindungen noch Acetat verwerten. Eine Ausnahme bildet der im Menschen verkommene M. stadtmanae, der Methanol und Wasserstoff zum Wachstum benötigt. Methanogenen ohne Cytochrome genügt ein H2-Partialdruck von unter 10 Pa, um die Methanogenese durchzuführen. Ihre Verdopplungszeit liegt bei unter einer Stunde pro Verdopplung. Unter den Methanogenen ohne Cytochromen findet man viele hyperthermophile Arten.
Biochemische Reaktionen
Bei der Reduktion von Carboxygruppen (–COOH) und von Kohlenstoffdioxid zu Methan spielen Enzyme mit charakteristischen Coenzymen eine wesentliche Rolle. Insbesondere sind dies die Coenzyme Tetrahydromethanopterin, Coenzym M, Coenzym F430 und F420, sowie spezielle Elektronen- beziehungsweise Wasserstoffüberträger. Diese kommen teilweise nur bei Methanbildnern vor. Für den komplexen biochemischen Vorgang sind über 200 Gene nötig, die für die entsprechenden Enzyme und Coenzyme kodieren.[7]
Methanogenese aus Kohlenstoffdioxid und Wasserstoff
Reduktion von Kohlenstoffdioxid zu Methan
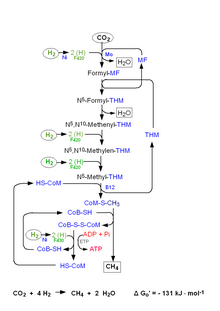

Übersicht EC-Nummern
- Formylmethanofuran-Dehydrogenase
EC
 1.2.99.5
1.2.99.5 - Methenyl-H4MPT-Cyclohydrolase EC
3.5.4.27
- Methylen-H4MPT-Dehydrogenase EC
1.5.99.9
- F420-abhängige Methylen-Reduktase
EC
1.5.99.11
- F420-unabhängige Methylen-Reduktase EC
1.12.98.2
- F420-reduzierende Hydrogenase EC
1.12.98.1
- Methyltransferase
EC
2.1.1.86
- Methyl-Coenzym-M-Reduktase
EC
2.8.4.1
- cytosolische Hydrogenase/Reduktase EC
1.8.98.1
.svg.png)
.svg.png)
Damit Kohlenstoffdioxid als Substrat genutzt werden kann, wird es zunächst an die eine reaktive Aminogruppe des Coenzyms Methanfuran (MFR) geknüpft. Dabei entsteht N-Carboxymethanofuran, ein instabiles Zwischenprodukt, das zum ersten stabilen Intermediat, dem N-Formylmethanofuran (CHO-MFR), reduziert wird. Eine Formylmethanofuran-Dehydrogenase (MFR-Dehydrogenase) katalysiert diese beiden Reaktionen und benötigt ein Reduktionsmittel in Form von reduziertem Ferredoxin. Die für diese Reduktion notwendigen Elektronen stammen dabei entweder aus Wasserstoff, die eine Hydrogenase auf oxidiertes Ferredoxin überträgt. Alternativ gelangen sie aus der Oxidation von Formiat, dem Anion der Ameisensäure, zu Kohlenstoffdioxid, was eine Formiat-Dehydrogenase katalysiert. Da die Bildung von CHO-MFR endergon ist, wird die nötige Energie aus dem elektrochemischen Ionengradienten der Membran angezapft.[2]
Die an MFR gebundene Formylgruppe (–CHO) wird auf Tetrahydromethanopterin (H4MPT) übertragen, das strukturell dem Tetrahydrofolat (THF) anderer Organismen ähnelt. Anschließend wird die an H4MPT gebundene Formylgruppe schrittweise über N5,N10-Methenyl-H4MPT und N5,N10-Methylen-H4MPT zu Methyl-H4MPT (–CH3) reduziert. Dieser Prozess ist vollständig reversibel und kann auch in entgegengesetzter Richtung ablaufen. Reduktionsmittel ist hier F420H2. Eine cytosolische Methenyl-H4MPT-Cyclohydrolase, Methylen-H4MPT-Dehydrogenase beziehungsweise eine (F420-abhängige) Methylen-Reduktase katalysieren diese Reaktionen. In Methanosarcinaarten liegt Tetrahydosarcinapterin (H4SPT) vor, das dem H4MPT sehr ähnlich ist.[2]
Neben der F420-abhängigen Methylen-Reduktase nutzen manche obligaten Hydrogenotrophe Wasserstoff direkt. Diese Methylen-Reduktase enthält im Gegensatz zu anderen Hydrogenasen weder Eisen-Schwefel- noch Nickel-Eisen-Cluster, sie ist „metallfrei“.
Das universelle Reduktionsmittel F420H2 wird nach Oxidation durch eine Eisen-Nickel enthaltende F420-reduzierende Hydrogenase regeneriert, welche Wasserstoff benötigt.
Die Methylgruppe aus Methyl-H4MPT wird dann auf das einfachste Coenzym, Coenzym M (CoM), übertragen. Es entsteht Methyl-CoM, bei der die Methylgruppe mit dem Sulfidrest des Coenzyms verknüpft ist (H3C–S-CoM). Der Transfer erfolgt über eine membrangebundene Methyltransferase. Diese Reaktion ist exergon (ΔG0'= −29 kJ/mol.[2]) Methanogene nutzen die hierbei freiwerdende Energie, um etwa zwei Natriumionen pro Umsetzung aus der Zelle zu exportieren. Dadurch bildet sich ein elektrochemisch wirkender Natriumionkonzentrationsunterschied.
Methyl-CoM reagiert schließlich mit Coenzym B (CoB) zu einem gemischten Disulfid, CoM-S–S-CoB, und Methan. Dies ist die Schlüsselreaktion der Methanogenese. Das gemischte Disulfid bezeichnet man auch als Heterodisulfid. Diese Reaktion wird von einer Methyl-Coenzym-M-Reduktase katalysiert, die den Cofaktor F430 enthält.
In der Bilanz wird damit ein Molekül Kohlenstoffdioxid umgesetzt gemäß:

Regeneration der Coenzyme M und B
Die Coenzyme M und B müssen für einen erneuten Durchgang regeneriert werden. Dies erfolgt durch eine Reduktion von CoM-S–S-CoB zu CoM und CoB und wird durch eine Heterodisulfidreduktase katalysiert. Die für diese Reaktion benötigten Elektronen entstammen entweder aus Wasserstoff, reduziertem Ferredoxin oder F420H2. Bei der Reaktion wird Energie freigesetzt (ΔG0'= −39 kJ·mol−1).
In Methanogenen mit Cytochromen wird CoM-S–S-CoB an einer membranständigen Heterodisulfidreduktase reduziert. In obligaten kohlenstoffreduzierenden Methanogenen ist dies ein Komplex mit drei Untereinheiten (HdrABC), in Methanosarcina-Arten ist er aus zwei Untereinheiten aufgebaut (HdrDE).[25] Für die Reduktion werden Elektronen benötigt. Entweder wird Wasserstoff an einer membrangebundenen Hydrogenase oxidiert, die unter anderem Häm b als prosthetische Gruppe enthält (Vho). Parallel dazu werden Protonen nach außen transportiert. Der Hydrogenasekomplex wurde beispielsweise im Süßwasser lebenden Ms. barkeri identifiziert. Ms. acetivorans, ein im Salzwasser vorkommendes Archaeon, oxidiert statt Wasserstoff Ferredoxin an einen ebenfalls membranständigen Komplex (Ma-Rnf), der u.a. Cytochrom c als prosthetische Gruppe aufweist. Dabei werden Natriumionen nach außen befördert. Falls Ms. acetivorans ausschließlich auf Kohlenmonoxid wächst, oxidiert ein membranständiger F420-Dehydrogenasekomplex (Fpo) reduziertes F420, bei dem Vorgang werden Protonen exportiert. Die Übertragung der Elektronen vom Hydrogenase- beziehungsweise Dehydrogenasekomplex zur Heterodisulfidreduktase wird durch Methanophenazin vermittelt. Die Reduktion von CoM-S–S-CoB ist exergon, daher werden durch diesen Prozess ebenfalls gleichzeitig Protonen nach außen transportiert, so dass insgesamt eine protonenmotorische Kraft aufgebaut wird. Diese nutzen die Methanogenen zum Aufbau von ATP (vgl. Abschnitt unten).
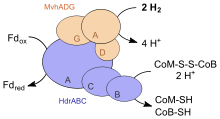
Dagegen besitzen Methanogene ohne Cytochrome weder Methanophenazin noch eine membrangebundene Heterodisulfidreduktase.[26] Für die Oxidation des Heterodisulfides CoM-S–S-CoB nutzen sie eine cytosolische Hydrogenase/Reduktase, die Wasserstoff benötigt und die freiwerdende Energie zur Reduktion von Ferredoxin koppelt. Jedoch liegt kein Mechanismus zugrunde, bei der die freiwerdende Energie zum Aufbau einer protonenmotorischen Kraft gekoppelt werden könnte – die Hetereosulfidreduktase liegt nicht membrangebunden vor. Daher können Methanogene ohne Cytochrome nur den Natriumionenkonzentrationsunterschied nutzen, der bei der Methyltransferasereaktion aufgebaut wird.
Umsetzung von Formiat zu Methan
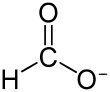
Ameisensäure bzw. sein Anion, Formiat (HCOO−), kann von etwa der Hälfte aller Methanogenen als Substrat genutzt werden.[16] Im Gegensatz zu Kohlenstoffdioxid wird es nicht direkt auf MFR übertragen, sondern zunächst durch eine Formiat-Dehydrogenase zu Kohlenstoffdioxid oxidiert. Das Enzym enthält Molybdän und Eisen-Schwefel-Cluster, es wurde bereits aus methanogenen Archaeen isoliert (beispielsweise aus Methanobacterium formicicium und Mc. vannielii). Bei der katalysierten Reaktion wird gleichzeitig F420 zu F420H2 reduziert. Kohlenstoffdioxid wird danach, wie weiter oben beschrieben, zu Methan reduziert.
Wie für die schrittweise Umsetzung von Kohlenstoffdioxid zu Methan werden auch für die Umsetzung von Formiat zu Methan an vier Stellen Reduktionsmittel benötigt. An zwei Stellen werden sie direkt in Form von F420H2 bei der stufenweisen Reduktion von Methenyl-H4MPT zu Methyl-H4MPT verbraucht. An den anderen beiden wird Wasserstoff für die cytosolische Heterodisulfidreduktase benötigt, das die Oxidation von CoM-S–S-CoB zu CoM und CoB und die Bildung von reduziertem Ferredoxin koppelt.[26] Wasserstoff kann entweder durch die F420-reduzierende Hydrogenase erzeugt werden oder alternativ durch eine Nickel-freie Hydrogenase.[27] Das bei der Heterodisulfidreduktase-Reaktion gebildete reduzierte Ferredoxin wird für die MFR-Dehydrogenase in der Eingangsreaktion benötigt.
Daher sind insgesamt vier Moleküle F420H2 erforderlich, um Formiat in der Methanogenese zu verwerten. Diese werden bereitgestellt, indem vier Moleküle Ameisensäure zu Kohlenstoffdioxid oxidiert werden. Drei Moleküle Kohlenstoffdioxid werden freigesetzt, und das vierte schließlich zu Methan umgesetzt. In der Bilanz ergibt sich damit:

Methanogenese mit methylierten C1-Verbindungen
C1-Verbindungen mit einer Methylgruppe wie beispielsweise Methylamin (CH3NH2) oder Methanol (CH3OH) kommen insbesondere in Meerwasser oder Brackwasser vor und sind anaerobe Abbauprodukte zellulärer Bestandteile bestimmter Pflanzen und des Phytoplanktons.[16]
Da der Kohlenstoff in der Methylgruppe bereits stärker reduziert ist als in CO2, müssen diese Verbindungen nicht den gesamten Weg wie der beim Kohlenstoffdioxid durchlaufen. Sie werden daher im unteren Drittel des Weges in der Methanogenese in Form von CH3–CoM eingespeist. Neben dem direkten Weg zu Methan werden methylierte Verbindungen auch zu Kohlenstoffdioxid oxidiert. Es gibt also einen oxidativen und einen reduktiven Zweig. Das liegt daran, dass die Elektronen für den reduktiven Zweig aus der Oxidation von Methylgruppe zu Kohlenstoffdioxid entnommen werden müssen, da die Nutzung von Wasserstoff aus der Umgebung (als Elektronenquelle) häufig nicht möglich ist.
Ein Molekül Methanol wird beispielsweise zu Kohlenstoffdioxid oxidiert, so dass mit Hilfe der freigesetzten Reduktionsäquivalente drei Moleküle zu Methan reduziert werden. Diese Disproportionierung erfolgt z.B. gemäß:

Diese beiden Zweige treten auch bei der Umsetzung von Methylaminen durch Methanosarcina auf. Methylamine werden zu Methan, CO2 und Ammoniak (NH3) verstoffwechselt, wobei drei der Methylgruppen zu Methan reduziert und eine zu Kohlenstoffdioxid oxidiert wird.
Hierbei wird die Methylgruppe des Substrates auf CoM übertragen und schließlich – wie oben beschrieben – zu Methan reduziert. Den Transfer auf CoM katalysieren cytosolische Methyltransferasen, die für die Reaktion Pyrrolysin als 22. Aminosäure benötigen und ein Corrinoid als prosthetische Gruppe enthalten.
Im oxidativen Zweig wird die Methylgruppe auf H4MPT durch eine membrangebundene Methyl-H4MPT-CoM-Methyltransferase übertragen. Da diese Reaktion Energie verbraucht (endergon ist), wird hierfür der elektrochemische Natriumionengradient angezapft. Methyl-H4MPT wird dann, in umgekehrter Reihenfolge wie oben beschrieben, zu Formyl-H4MPT oxidiert, wobei gleichzeitig F420 reduziert wird. Die Formylgruppe wird an MFR gekoppelt und schließlich durch die Formyl-Dehydrogenase zu Kohlenstoffdioxid oxidiert. Formal entsprechen also die Reaktionen des oxidativen Zweiges der umgekehrten Verstoffwechslung von Kohlenstoffdioxid zu CH3-CoM.
Beispielsweise werden vier Moleküle Methylamin umgesetzt zu:

Allgemein werden methylierte C1-Verbindungen abgebaut gemäß:

(mit R = –SH, –OH, –NH2, –NHCH3, –N(CH3)2, –N(CH3)3+)
Spaltung von Acetat zu Methan und Kohlenstoffdioxid
Übersicht EC-Nummern
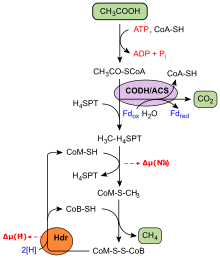
Acetat (CH3COOH) ist die einzige C2-Verbindung für die Methanogenese, die nur Vertreter der Gattungen Methanosaeta und Methanosarcina umsetzen können. Im Vergleich zu allen anderen Methanbildnern stammt indes der überwiegend größere Teil an Methan weltweit aus der Spaltung von Acetat.[16]
Um als Substrat für die Methanogenese genutzt zu werden, wird Acetat zunächst „aktiviert“. Dies erfolgt dadurch, dass es an Coenzym A verknüpft wird, so dass Acetyl-CoA entsteht. Hierbei wurden zwei Stoffwechselwege identifiziert:
- Entweder geschieht die Aktivierung direkt durch eine Acetyl-CoA-Synthetase, bei dem Vorgang wird ein Molekül ATP zu AMP und Pyrophosphat (PPi) gespalten. Die Acetyl-CoA-Synthetase findet man in obligat acetotrophen Methanogenen der Gattung Methanosaeta.
- Alternativ erfolgt der Prozess schrittweise: Acetat wird durch eine Acetatkinase mittels ATP zunächst phosphoryliert, dabei entsteht Acetylphosphat.[28] Dieses reagiert mit Coenzym A zu Acetyl-CoA. Eine Phosphotransacetylase katalysiert die zweite Reaktion.
Acetyl-CoA (CH3-CO-SCoA) wird für den weiteren Verlauf in drei Bestandteile gespalten: Coenzym A (HS-CoA), die Methylgruppe (–CH3) und die Carboxygruppe (–CO). Diese Reaktion findet im CO-Dehydrogenase/Acetyl-CoA-Synthase-Komplex (kurz CODH/ACS) statt. Der Komplex transferiert die Methylgruppe auf H4MPT, das wie weiter oben beschrieben zu Methan umgesetzt wird. CO wird enzymgebunden zu CO2 oxidiert, die dabei freiwerdenden Elektronen gelangen auf Ferredoxin, das für die Regenerierung von Coenzym B und M benötigt wird. Die Spaltung von Acetyl-CoA in drei Bestandteile entspricht formal der Umkehrung des reduktiven CoA-Weges, bei der Acetyl-CoA gebildet wird. Aus einem Molekül Acetat wird somit ein Molekül Kohlenstoffdioxid sowie ein Molekül Methan gebildet, gemäß:

Energiegewinnung
ATP-Synthese
Im Zuge der Methanogenese wird sowohl ein Protonen-, als auch ein Natriumionen-Konzentrationsunterschied erzeugt, was gleichzeitig zu einer Energetisierung der Zellmembran führt (ΔµH+, ΔµNa+).[2] Dabei sind Methanogene die einzigen Organismen, die diese beiden Konzentrationsunterschiede parallel aufbauen. Wie bei der anaeroben oder aeroben Atmung wird die Energie beider Konzentrationsunterschiede zum Aufbau von ATP durch eine ATP-Synthase genutzt.
Archaeen besitzen ATP-Synthasen des Typs A1AO, Bakterien, Mitochondrien und Chloroplasten die F1FO-ATP-Synthase und Eukaryoten die vom Typ V1VO. Hierbei nutzen Methanogene eine A1AO-ATP-Synthase. Im Genom von Ms. barkeri und Ms. acetivorans wurden zwar auch Gene für eine bakterielle F1FO-ATP-Synthase entdeckt. Jedoch ist es nicht einmal sicher, ob diese auch abgelesen werden und überhaupt funktionell vollständig sind.[2] Wahrscheinlich sind diese Gene durch horizontalen Gentransfer in das Genom jener Archaeen gelangt.
Ob die A1AO-ATP-Synthase in methanogenen Archaeen sowohl Protonen als auch Natriumionen akzeptiert, ist noch nicht eindeutig geklärt. Durch das Vorhandensein eines Na+/H+-Antiporters kann der elektrochemische Natriumionen-Konzentrationsunterschied aber jederzeit in eine protonenmotorische Kraft umgewandelt werden. So hat man im Genom von Ms. mazei drei dieser Transporter identifiziert.
Die genaue Struktur der ATP-Synthase ist noch Gegenstand der Forschung. A1AO-ATP-Synthasen ähneln zwar eukaryotischen des Typs V1VO, sind aber funktionell anders – sie erzeugen ATP, während letztere ATP zum Aufbau eines Ionengradienten hydrolysieren und damit verbrauchen.[16] Die meisten Archaeen haben einen Rotor von 12 Gruppen. Die katalytische Domäne, an der ATP erzeugt wird, weist drei Bindestellen auf. Damit genügen vier Protonen zur Synthese eines Moleküls ATP. Als Ausnahme gilt die ATP-Synthase in Mc. janaschii und Mc. maripaludis, bei denen das Rotorelement nur über 8 Gruppen verfügt. Damit genügen durchschnittlich 2,6 Protonen für die Synthese eines Moleküls ATP.
Energieausbeute
Die Reduktion von Kohlenstoffdioxid zu Methan durch Wasserstoff ist exergon (energiefreisetzend). Unter Standardbedingungen bei pH 7 beträgt die Änderung der Gibbs-Energie ΔG0’ je nach Literaturangabe −130,[17] −131[2][9][20] oder −135[4] kJ/mol CH4. Unter solchen Bedingungen würden in der Methanogenese je Molekül gebildeten Methans drei Moleküle ATP aus ADP und Pi gebildet werden können. Die ΔG0’-Werte bei den anderen methanogenen Reaktionen sind in obiger Tabelle aufgeführt.
Für die Berechnung von ΔG0’ werden – neben einer Temperatur von 25 °C und einem pH-Wert von 7 – Konzentrationen der gelösten Gase im Gleichgewicht mit Gasdrücken von 105 Pa vorausgesetzt.[9] Dies entspricht jedoch nicht den natürlichen Bedingungen, denn solche hohen Gaskonzentrationen kommen in den Habitaten weder vor, noch könnten sie in der Zelle aufrechterhalten werden. Damit fällt die Energieausbeute unter natürlichen Bedingungen niedriger aus.
In den meisten Habitaten herrscht ein H2-Gasdruck von etwa 1–10 Pa vor.[9] Unter diesen Bedingungen und pH=7 liegt die Änderung der Freien Energie (ΔG) bei etwa −17 bis −40 kJ/mol Methan, womit weniger als durchschnittlich ein Molekül ATP pro erzeugtem Molekül Methan gebildet werden kann. Außerdem spielen für die Berechnung von ΔG der pH-Wert, der vorherrschende Druck und auch die Temperatur eine Rolle. So fällt die Änderung der Freien Energie bei der Reduktion von Kohlenstoffdioxid zu Methan durch Wasserstoff unter Standardbedingungen (25 °C) von −131 kJ/mol auf −100 kJ/mol, wenn eine Temperatur von 100 °C vorliegt.[9]
Auch bei der Verwendung anderer C1-Verbindungen ist ΔG' gering, so dass viele Methanogene knapp am „thermodynamischen Limit“ wachsen.[2]
Evolution
Genomische Analysen zeigen, dass sich die Methanogenese früh in Euryarchaeota und erst nach Abspaltung der Thermococcales etabliert hatte.[29] Dies wird dadurch untermauert, dass alle Methanogenen die gleichen homologen Enzyme und Cofaktoren für den zentralen methanogenen Stoffwechselweg teilen. Darüber hinaus ist die Methanogenese in der Evolution wahrscheinlich nur einmal aufgetreten, da sich ein horizontaler Gentransfer zwischen den Methanogenen nicht nachweisen lässt. So liegen zwischen den Ordnungen Methanopyrales, Methanobacteriales, Methanococcales (Klasse I-Methanogene) sowie Methanomicrobiales (Klasse II-Methanogene) und Methanosarcinales (Klasse III-Methanogene) Ordnungen, die keine Methanogenese durchführen können, z.B. die Thermoplasmatales, Archaeoglobales und Halobacteriales. Zwar können beispielsweise in A. fulgidus noch Enzyme für die ersten Schritte der Methanogenese nachgewiesen werden. Dem Archaeon fehlen Enzyme für die letzten beiden Schritte, so auch die Coenzym M-Reduktase. Wahrscheinlich haben die Archaeen in diesen drei Ordnungen die Fähigkeit zur Methanogenese im Laufe der Evolution unabhängig voneinander verloren.
Warum recht früh und „plötzlich“ die Methanogenese in Euryarchaeota aufgetreten ist, bleibt noch Gegenstand der Forschung. Über die Entstehung der Methanogenese gibt es verschiedene Theorien. Eine davon besagt, dass der letzte gemeinsame Vorfahre aller Archaeen selbst ein methanogener Organismus war.[29] Manche Archaeen betreiben Methanogenese in Umgebungen extremen Salz- und Säuregehaltes sowie hoher Temperaturen. Da gerade diese Umweltbedingungen vermutlich auch nach der Entstehung der Erde vorgeherrscht haben, könnten methanogene Archaeen zu den ersten Lebensformen gezählt haben.[2] Demzufolge müsste aber die Fähigkeit zur Methanogenese sowohl in allen Crenarchaeota als auch in allen anderen nicht-methanogenen Linien unabhängig voneinander verloren gegangen sein, was als recht unwahrscheinlich angesehen wird.[21]
Nach einer anderen Theorie liegt der Ursprung der Methanogenese möglicherweise in der Oxidation von Methan, also im umgekehrten Stoffwechselweg. Diese auch methanotroph genannten Organismen oxidieren Methan zu Kohlenstoffdioxid und Wasser, wobei dies in Bakterien aerob und Archaeen anaerob[30] geschieht. Dagegen spricht allerdings eine gegenteilige Annahme: Diese besagt, dass solche methanotrophen Archaeen eher aus methanogenen Archaeen hervorgegangen sind. Denn es wurde postuliert, dass die Methanogenese, die anaerobe Methanotrophie der Archaeen und die aerobe Methanotrophie der Bakterien aus einem gemeinsamen Stoffwechselweg hervorgegangen sind, der im letzten gemeinsamen Vorfahren (MCRA, engl. für most recent common ancestor) ursprünglich zur Entgiftung von Formaldehyd diente.
Eine neue Theorie betrachtet die Rolle Pyrrolysins (Pyl) im Methyl-Corrinoid-Weg der Methanosarcinales, durch den Methylamine in die Methanogenese eintreten können.[21] Die Methylgruppe dieser Methylamine wird durch eine spezifische Methyltransferase auf ein Corrinoid-enthaltenes Protein übertragen (vgl. Abschnitt oben). Methyltransferasen enthalten die 22. Aminosäure Pyrrolysin im katalytisch aktiven Zentrum. Pyrrolysin wurde so gut wie in keinem anderen Enzym nachgewiesen. Da die gesamte Pyl-Maschinerie stammesgeschichtlich als sehr alt gilt, vermutet man, dass sie durch horizontalen Gentransfer aus vermutlich mehreren Donorlinien stammt, die entweder inzwischen alle ausgestorben sind oder noch nicht entdeckt wurden. Dies setzt aber auch voraus, dass die Donorlinie, aus der die Pyl-Maschinerie stammt, bereits einen gewissen Grad an Diversität zu dem Zeitpunkt erreicht hatte, als noch ein gemeinsamer Vorfahre unserer drei Domänen existierte.
Nur in Methanosarcinales wurden Cytochrome gefunden. Sie verfügen über ein breiteres Substratspektrum als Methanogene ohne Cytochrome, sie nutzen beispielsweise auch Acetat. Man nimmt an, dass sich die Methanogenese aus Acetat erst spät entwickelt hat. Vermutlich sind die für die Acetat-Nutzung benötigten Gene der Acetat-Kinase erst durch horizontalen Gentransfervon einem Cellulose abbauenden, zu den Clostridien gehörenden acetogenenBakterium in die methanogenen Archaea gelangt.[31][32]
Bei Wachstum auf Kohlenstoffdioxid + Wasserstoff benötigen Methanosarcinales hohe H2-Konzentrationen, so dass sie bei geringeren Gasdrücken immer von Methanogenen ohne Cytochrome übervorteilt werden. Dies führte im Laufe der Evolution dazu, dass manche Methanosarcinales, wie beispielsweise Ms. acetivorans, Methanolobus tindarius und Methanothrix soehngenii, vollständig die Fähigkeit verloren haben, Kohlenstoffdioxid als Substrat unter Mitverwendung von Wasserstoff zu nutzen.[9] Da die Methanogenese mit Kohlenstoffdioxid und Wasserstoff sehr weit verbreitet ist, geht man davon aus, dass diese Form die ursprünglichste ist.[6]
Andere Arten der biologischen Methanfreisetzung
Auch unter aeroben Bedingungen wurde eine biogene Methanfreisetzung beobachtet. 2006 wurde postuliert, dass lebende Pflanzen und totes Pflanzenmaterial bis zu 40 % zur globalen biologisch erzeugten Methanmenge beitragen.[33] Dies wurde durch spätere Messungen aber revidiert, aus denen hervorging, dass Pflanzen nur einen vergleichsweise sehr geringen Teil Methan produzieren.[34] Darüber hinaus scheint es sich hierbei nicht um einen spezifischen Stoffwechselweg zu handeln. Stattdessen führt beispielsweise hoher UV-Stress zur spontanen Zerstörung von Biomasse, wodurch Methan gebildet wird. Außerdem könnte in Wasser gelöstes Methan in der Pflanze freigesetzt und in die Atmosphäre abgegeben werden.[35]
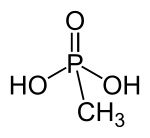
Auch für marine Mikroorganismen wie Bakterien wurde eine aerobe Methanogenese postuliert. Diese können Methylphosphonsäure (MPS) durch eine spezielle Lyase zu Phosphonat und Methan spalten.[36] Jedoch wurde MPS weder frei in marinen Ökosystemen nachgewiesen, noch ist es eine natürlich vorkommende Verbindung.[37] Eine mögliche Quelle für Methylphosphonsäure könnte das Archaeon Nitrosopumilus maritimus sein, das Polysaccharide erzeugt, die mit MPS verknüpft sind und einen Stoffwechselweg aufweist, der Phosphoenolpyruvat zu MPS umsetzen kann.
In-vitro wurde ein neuer enzymatischer Mechanismus für eine bakterielle SAM-abhängige Lyase gezeigt, der Ribose-1-phosphonat-5-phosphat zu Methan und Ribose-1,2-zyklisches Phosphat-5-phosphat spaltet.[38] Wenn die Konzentration von Phosphonaten niedrig ist, können mit der Lyase unreaktive Kohlenstoff-Phosphor-Verbindungen unter aeroben Bedingungen gespalten werden; dabei wird Methan freigesetzt.
Möglicherweise setzen auch saprotrophe Pilze stoffwechselspezifisch Methan aus Methionin frei.[39]
Das Bakterium Rhodopseudomonas palustris kann mittels einer reinen Eisen-haltigen Nitrogenase in einer einzigen enzymatischen Reaktion N2 zusammen mit CO2 und Protonen zu Methan, Ammoniak und H2 umsetzen.[40]
Anwendung

Das aus Biomasse mikrobiell gebildete Methan enthält einen großen Teil der Energie, die im Ausgangsprodukt gespeichert war. Das macht man sich in verschiedenen technischen Anwendungen zu Nutze. So wird in Fermentern von Biogasanlagen, Faultürmen von Klärwerken und in Deponiekörpern die Methanbildung zur Erzeugung von Faulgasen (Biogas, Klärgas, Deponiegas) verwendet. Die dabei eingesetzte Biomasse wäre mit anderen Verfahren nicht oder nur schwierig energetisch nutzbar.
Die Nutzung des Methans in technischen Anwendungen, wie z.B. einem an eine Biogasanlage angeschlossenes Blockheizkraftwerk (BHKW), erfolgt durch Oxidation mit Sauerstoff:

Literatur
- Lexikon der Biologie. Band 9, Spektrum Akademischer Verlag, Heidelberg 2002, ISBN 3-8274-0334-0.
- Georg Fuchs (Hrsg.): Allgemeine Mikrobiologie. Begründet von Hans-Günter Schlegel, 8. Auflage. Georg Thieme Verlag, Stuttgart, New York 2007, ISBN 978-3-13-444608-1.
- Michael T. Madigan, John M. Martinko, Paul V. Dunlap, David P. Clark: Brock – Biology of Microorganisms. 12. Auflage. Pearson, San Francisco 2009, ISBN 0-321-53615-0.
- Rudolf K. Thauer, Anne Kristin Kaster, Meike Goenrich, Michael Schick, Takeshi Hiromoto, Seigo Shima: Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2
storage. In: Annual Review of Biochemistry. Band 79, 2010.
PMID 20235826,
doi:10.1146/annurev.biochem.030508.152103,
S. 507–536
- G. Fournier: Horizontal gene transfer and the evolution of methanogenic pathways. In: Methods in Molecular Biology. Band 532, 2009.
PMID 19271184,
doi:10.1007/978-1-60327-853-9, S. 163–179
- Rudolf K. Thauer, Anne Kristin Kaster, Henning Seedorf, Wolfgang Buckel, Reiner Hedderich: Methanogenic archaea: ecologically relevant differences in energy conservation. In:
Nature Reviews Microbiology. Band 6, Nr. 8, 2008.
PMID 18587410,
doi:10.1038/nrmicro1931, S. 579–591
- U. Deppenmeier, V. Müller: Life close to the thermodynamic limit: how methanogenic archaea conserve energy. In: Results and Problems in Cell Differentiation.
Band 45, 2008. PMID 17713742,
doi: 10.1007/400_2006_026, S. 123–152
- J. G, Ferry (): How to make a living by exhaling methane. In: Annual Review of Microbiology. Band 64, 2010.
PMID 20528692,
doi:10.1146/annurev.micro.112408.134051,
S. 453–473
Einzelnachweise
- ↑ Michael T. Madigan, John M. Martinko: Brock – Mikrobiologie. 11. überarbeitete Auflage. Übersetzung von Brock – Biology of microorganisms 11. ed. ins Deutsche. Pearson Studium, München 2006, ISBN 3-8273-7187-2.
- ↑ Hochspringen nach: a
b c d
e f g
h i j
U. Deppenmeier, V. Müller: Life close to the thermodynamic limit: how methanogenic archaea conserve energy. In: Results and Problems in Cell Differentiation.
Band 45, 2008.
PMID 17713742,
doi: 10.1007/400_2006_026, S. 123–152.
- ↑ Georg Fuchs (Hrsg.): Allgemeine Mikrobiologie, begründet von Hans-Günter Schlegel. 8. Auflage. Georg Thieme Verlag, Stuttgart, New York 2007, ISBN 978-3-13-444608-1, S. 397.
- ↑ Hochspringen nach: a b
c d e f
g h
Y. Liu, W. B. Whitman: Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. In: Annals of the New York Academy of Sciences. Band 1125, 2008.
PMID 18378594,
doi:10.1196/annals.1419.019, S. 171–189.
- ↑ Joan L. Slonczewski, John W. Foster: Mikrobiologie: Eine Wissenschaft mit Zukunft. 2. Auflage. Spektrum Akademischer Verlag, Berlin, Heidelberg 2012, ISBN 978-3-8274-2909-4, S. 854.
- ↑ Hochspringen nach: a b
c d e
f Franziska Enzmann et al.: Methanogens: biochemical background and biotechnological applications.
In: AMB Express. Band 8,
Nr. 1, 4. Januar 2018,
doi: 10.1186/s13568-017-0531-x,
PMID 29302756,
PMC 5754280 (freier Volltext).
- ↑ Hochspringen nach: a
b c
d e
f g Zhe Lyu, Nana Shao, Taiwo Akinyemi, William B. Whitman:
Methanogenesis. In: Current Biology.
Band 28,
Nr. 13, 9. Juli 2018, ISSN 0960-9822,
S. R727–R732,
doi: 10.1016/j.cub.2018.05.021,
PMID 29990451.
- ↑ R. K. Dhaked, P. Singh, L. Singh: Biomethanation under psychrophilic conditions. In: Waste Manag. Band 30 (12), 2010, S. 2490–2496. PMID 2072413, doi:10.1016/j.wasman.2010.07.015.
- ↑ Hochspringen nach: a b
c d e
f g h
Rudolf K. Thauer, Anne Kristin Kaster, Henning Seedorf, Wolfgang Buckel, Reiner Hedderich: Methanogenic archaea: ecologically relevant differences in energy conservation. In:
Nature Reviews Microbiology. Band 6, Nr. 8, 2008, PMID 18587410,
doi:10.1038/nrmicro1931, S. 579–591.
- ↑ Gerhard Gottschalk: Welt der Bakterien, Archaeen und Viren: Ein einführendes Lehrbuch der Mikrobiologie. 1. Auflage. John Wiley & Sons, 2015, ISBN 978-3-527-68892-0, S. 114.
- ↑
Sanae Sakai, Hiroyuki Imachi, Satoshi Hanada, Akiyoshi Ohashi, Hideki Harada: Methanocella paludicola gen. nov., sp. nov., a methane-producing
archaeon, the first isolate of the lineage 'Rice Cluster I', and proposal of the new archaeal order Methanocellales ord. nov. In:
International Journal of Systematic and Evolutionary Microbiology.
Band 58, Pt 4, April 2008, ISSN 1466-5026,
S. 929–936,
doi: 10.1099/ijs.0.65571-0,
PMID 18398197.
- ↑
S. Sakai et al.: Methanocella arvoryzae sp. nov., a hydrogenotrophic methanogen isolated from rice field soil. In: International Journal of Systematic and Evolutionary
Microbiology, Band 60(Pt 12), 2010, S. 2918–2923. PMID 20097796,
doi:10.1099/ijs.0.020883-0.
- ↑ K. Paul et al.: 'Methanoplasmatales': Thermoplasmatales-related archaea in termite guts and other
environments are the seventh order of methanogens. In: Applied and Environmental Microbiology, 2012,
PMID 23001661,
doi:10.1128/AEM.02193-12.
- ↑ Beschreibung: Diversity, ultrastructure, and comparative genomics of “Methanoplasmatales”, the seventh order of
methanogens..
- ↑
I. Anderson et al.: Genomic characterization of methanomicrobiales reveals three classes of methanogens. In: PLoS One, Band 4 (6), 2009, S. e5797;
PMID 19495416,
doi:10.1371/journal.pone.0005797.
- ↑ Hochspringen nach: a b
c d e
f
U. Deppenmeier: The unique biochemistry of methanogenesis. In: Progress in Nucleic Acid Research and Molecular Biology. Band 71, 2002.
PMID 12102556,
doi:10.1016/S0079-6603(02)71045-3, S. 223–283
- ↑ Hochspringen nach: a b
U. Deppenmeier: Redox-driven proton translocation in methanogenic Archaea. In: Cellular and Molecular Life Sciences. Band 59 (9), 2002.
PMID 12440773,
doi:10.1007/s00018-002-8526-3, S. 1513–1533.
- ↑
WF. Fricke et al.: The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP
synthesis. In: J Bacteriol., 2006, 188(2), S. 642–658;
PMID 16385054;
PMC 1347301 (freier Volltext).
- ↑
E. Oelgeschläger, M. Rother: Carbon monoxide-dependent energy metabolism in anaerobic bacteria and archaea. In: Archives of Microbiology, Band 190(3), 2008.
PMID 18575848,
doi:10.1007/s00203-008-0382-6, S. 257–269.
- ↑ Hochspringen nach: a b
W. Martin, MJ. Russell: On the origin of biochemistry at an alkaline hydrothermal vent. In: Philos Trans R Soc Lond B Biol Sci., 2007, 362(1486), S. 1887–1925;
PMID 17255002;
PMC 2442388 (freier Volltext).
- ↑ Hochspringen nach: a b
c
G. Fournier: Horizontal gene transfer and the evolution of methanogenic pathways. In: Methods Mol Biol., 2009, 532; 163–179;
PMID 19271184;
doi: 10.1007/978-1-60327-853-9_9.
- ↑ Hochspringen nach: a b
AJ. Watkins et al.: Glycine betaine as a direct substrate for methanogens (Methanococcoides spp.). In: Appl Environ Microbiol., 2014, 80(1), S. 289–293;
PMID 24162571;
doi:10.1128/AEM.03076-13;
aem.asm.org (PDF).
- ↑ Hochspringen nach: a b
c
AJ. Watkins et al.: Choline and N,N-dimethylethanolamine as direct substrates for methanogens. In: Appl Environ Microbiol., 2012, 78(23), S. 8298–8303;
PMID 23001649;
doi:10.1128/AEM.01941-12;
aem.asm.org (PDF).
- ↑ z.B. Methanosphaera stadtmanae, ein im menschlichen Verdauungstrakt vorkommendes Archaeon, dessen Genom sequenziert wurde.
- ↑
JG. Ferry: How to make a living by exhaling methane. In: Annu Rev Microbiol., 2010, 64, S. 453–473;
PMID 20528692;
doi:10.1146/annurev.micro.112408.134051
- ↑ Hochspringen nach: a b
Rudolf K. Thauer, Anne Kristin Kaster, Meike Goenrich, Michael Schick, Takeshi Hiromoto, Seigo Shima: Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage.
In: Annual Review of Biochemistry, Band. 79, 2010, S. 507–536,
PMID 20235826,
doi:10.1146/annurev.biochem.030508.152103.
- ↑
B. Lupa et al.: Formate-dependent H2 production by the mesophilic methanogen Methanococcus maripaludis. In: Applied and Environmental Microbiology, 2008, Band 74, Nr. 21,
2008, S. 6584–6590 (englisch); PMID 18791018;
aem.asm.org (PDF).
- ↑
Acetylphosphat, Lexikon der Biologie;
Acetylphosphat, Lexikon der Chemie. Auf
spektrum.de.
- ↑ Hochspringen nach: a b
S. Gribaldo, C. Brochier-Armanet: The origin and evolution of Archaea: a state of the art. In: Philosophical Transactions of the Royal Society B: Biological Sciences.
Band 361 (1470), 2006, S. 1007–1022. PMID 16754611,
PMC 1578729 (freier Volltext).
- ↑
Martin Kruger, Anke Meyerdierks, Frank Oliver Glockner, Rudolf Amann, Friedrich Widdel, Michael Kube, Richard Reinhardt, Jorg Kahnt, Reinhard Bocher, Rudolf K. Thauer, Seigo Shima:
A conspicuous nickel protein in microbial mats that oxidize methane anaerobically. In:
Nature. 426. Jahrgang, Nr. 6968, 2003,
S. 878–881,
doi: 10.1038/nature02207.
- ↑ Gregory P. Fournier, J. Peter Gogarten: Evolution of acetoclastic methanogenesis in Methanosarcina via horizontal gene transfer from cellulolytic Clostridia. In: Journal of bacteriology. 190. Jahrgang, Nr. 3, 2008, S. 1124–1127.
- ↑
Sofya K. Garushyants, Marat D. Kazanov, Mikhail S. Gelfand: Horizontal gene transfer and genome evolution in Methanosarcina. In:
BMC Evolutionary Biology. 15. Jahrgang,
Nr. 1, 2015,
S. 1–14,
doi: 10.1186/s12862-015-0393-2.
- ↑
F. Keppler et al.: Methane emissions from terrestrial plants under aerobic conditions. In: Nature, 2006, 439(7073), S. 187–191;
PMID 16407949;
doi:10.1038/nature04420.
- ↑
TA. Dueck et al.: No evidence for substantial aerobic methane emission by terrestrial plants: a 13C-labelling approach. In: New Phytol., 2007, 175(1), S. 29–35 (englisch);
PMID 17547664;
doi:10.1111/j.1469-8137.2007.02103.x.
- ↑
RE. Nisbet et al.: Emission of methane from plants. In: Proc Biol Sci, 2009, 276(1660), S. 1347–1354 (englisch);
PMID 19141418;
royalsocietypublishing.org (PDF).
- ↑
DM. Karl et al.: Aerobic production of methane in the sea. In: Nature Geoscience, 2008, 1, S. 473–478;
doi:10.1038/ngeo234.
- ↑
WW. Metcalf: Synthesis of methylphosphonic acid by marine microbes: a source for methane in the aerobic ocean. In:
Science, 2012, 337(6098), S. 1104–1107; PMID 22936780;
doi:10.1126/science.1219875.
- ↑
SS. Kamat et al.: The catalytic mechanism for aerobic formation of methane by bacteria. In: Nature, 2013, 497(7447), S. 132–136;
PMID 23615610;
doi:10.1038/nature12061.
- ↑
K. Lenhart et al.: Evidence for methane production by saprotrophic fungi. In: Nat Commun, 2012, 3; 1046;
PMID 22948828;
doi:10.1038/ncomms2049.
- ↑ Yanning Zheng et al.: A pathway for biological methane production using bacterial
iron-only nitrogenase. In: Nature Microbiology.
Band 3,
Nr. 3, März 2018,
S. 281–286,
doi: 10.1038/s41564-017-0091-5.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 28.06. 2026