Peroxisom
| Übergeordnet |
| Organell |
| Untergeordnet |
| Membran Lumen Matrix Proteinkomplexe |
| Gene Ontology |
|---|


1. Nucleolus (Kernkörperchen)
2. Zellkern (Nukleus)
3. Ribosomen
4. Vesikel
5. Raues (Granuläres) ER (Ergastoplasma)
6. Golgi-Apparat
7. Cytoskelett
8. Glattes (Agranuläres) ER
9. Mitochondrien
10. Lysosom
11. Cytoplasma (mit Cytosol und Cytoskelett)
12. Peroxisomen
13. Zentriolen
14. Zellmembran
Peroxisomen, auch Microbodies (veraltet) genannt, sind Zellorganellen in eukaryotischen Zellen, die von einer Biomembran umgeben sind. Sie verbrauchen in vielfältigen Stoffwechselfunktionen Sauerstoff und gelten daher als die ersten Entgiftungsapparate, die mit dem Auftreten einer sauerstoffhaltigen Erdatmosphäre erforderlich wurden.[1]
Struktureller Aufbau
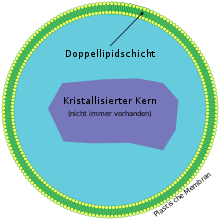
Es handelt sich bei Peroxisomen um kleine (100 – 1000 nm Durchmesser), mit einer einfachen Membran umhüllte Vesikel, die sich im Cytoplasma einer Zelle befinden.[2][3] In diesen räumlich abgetrennten Bereichen (Zellkompartimenten) können, durch die Membran geschützt, Reaktionen ablaufen, die für die Zelle gefährlich wären, würden sie im Cytoplasma erfolgen. Dies ist ein Beispiel für die Wichtigkeit der Zellkompartimentierung. Peroxisomen enthalten Enzyme für den Stoffwechsel von Wasserstoffperoxid (H2O2), weshalb sich der Begriff „Peroxisom“ etablierte. Morphologisch wurden sie früher auch als „Mikrobodies“ bezeichnet.
Anzahl, Größe und Proteinausstattung der Peroxisomen sind abhängig von Zelltyp und Wachstumsbedingungen. So hat man beispielsweise in Backhefe (S. cerevisiae) die Beobachtung gemacht, dass bei guter Glucoseversorgung nur einige wenige, kleine Peroxisomen vorhanden sind. Wenn dagegen die Hefen mit langkettigen Fettsäuren versorgt wurden, bildeten sich 20 bis 25 große Organellen.[4]
Häufig dient molekularer Sauerstoff als Co-Substrat, aus dem dann Wasserstoffperoxid (H2O2) gebildet wird. Der Wasserstoffperoxid-abbauenden Peroxidase verdanken die Peroxisomen ihren Namen.
Funktionen

In den Peroxisomen befinden sich ca. 60 Monooxygenasen und Oxidasen genannte Enzyme, die den oxidativen Abbau von Fettsäuren, Ethanol und anderen Verbindungen katalysieren. Diese Enzyme verwenden molekularen Sauerstoff als Co-Substrat, so dass sich für die Zellfunktion Wasserstoffperoxid bildet. Wasserstoffperoxid ist ein Zellgift im Cytoplasma und kann viele wichtige Biomoleküle zerstören.
Wasserstoffperoxid kann durch zwei Arten abgebaut werden. Eine Möglichkeit zur Entgiftung besteht in dessen sofortiger Umsetzung durch Katalase in einer Disproportionierungsreaktion, wobei Wasser und Sauerstoff entsteht:

Peroxisomen besitzen auch die namensgebende Peroxidase. Für ihre Funktion wird das Wasserstoffperoxid verbraucht, gemäß:

Oft sind die Enzymkonzentrationen so hoch, dass sie kristalline Aggregate (Nucleoide) bilden.
Nach der Endosymbiontentheorie wurden im weiteren Verlauf der Evolution Bakterien (vermutlich α-Proteobakterien) durch die „Urkaryoten-“ (Vorläufer der Eukaryoten, vermutlich Archaeen der Asgard-Gruppe, vgl. Eozyten-Hypothese) Zellen aufgenommen, die bereits über einen sinnvollen Sauerstoffverwertungsapparat (Citratzyklus nebst Atmungskette) verfügten und damit zur ATP-Synthese auf dem Wege der oxidativen Phosphorylierung befähigt waren. Dies waren die Vorläufer der „modernen“ Mitochondrien.
Die Peroxisomen wurden dabei nicht überflüssig, sondern sie wurden in den Katabolismus (Energiegewinn) eingebunden; zum Bindeglied wurde das (energiereiche) Acetyl-CoA. Die Abbildung zeigt beispielhaft, wie Ethanol eingesetzt wird, um nicht nur Wasserstoffperoxid zu entgiften, sondern auch selbst in einen Metaboliten (Acetyl-CoA) von allgemeiner Bedeutung im Katabolismus und Anabolismus (Aufbau von Fettsäuren, Cholesterin usw.) überführt zu werden. Peroxisomen tragen somit zur Verstoffwechselung von Ethanol bei.
Darüber hinaus katalysieren sie wichtige Schritte bei der Biosynthese von Lipiden (Plasmalogene) der Myelinscheide von Nerven (daher gehen Störungen ihrer Funktion oft mit neurologischen Schäden einher). Die konkreten Stoffwechselwege, die ausschließlich in Peroxisomen ablaufen, sind[5]
- die α-Oxidation von Phytansäure
- die β-Oxidation sehr langkettiger, mehrfach ungesättigter Fettsäuren
- die Biosynthese von Plasmalogenen
- die Konjugation von Cholsäure im Rahmen der Gallensäuresynthese
Andere Formen
Glyoxysomen (auch Glyoxisomen) sind spezialisierte Peroxisomen, die man im Endosperm und den Speichergeweben fettreicher Samenzellen findet. Sie erhielten ihren Namen, da sie am Glyoxylatzyklus beteiligt sind. Die in den Glyoxysomen enthaltenen Enzyme ermöglichen die Nutzung von Fetten zum Aufbau von Biopolymeren (Zucker, Proteine), die für das Pflanzenwachstum nötig sind.
In photosynthetisch aktiven Pflanzen nehmen Peroxisomen auch an der Photorespiration teil – dort ebenfalls in Kooperation mit Mitochondrien. Sie werden als Blatt-Peroxisomen bezeichnet. Pflanzliche Glyoxysomen und Blatt-Peroxisomen können sich ineinander umwandeln.[3]
Entstehung
Die Herkunft der Peroxisomen war in den letzten Jahren kontrovers diskutiert. Heutzutage ist bekannt, dass sich Peroxisomen analog zu Mitochondrien durch Teilung innerhalb der Zelle vermehren können. Die de novo-Bildung neuer Peroxisomen ist ein mehrstufiger Prozess, der mit der Abschnürung von Vorläufervesikeln aus dem Endoplasmatischen Retikulum (ER) beginnt. Wahrscheinlich fusionieren dann die kleinen Vorläuferversikel zu einem reifen Peroxisom. Für die Biogenese ist Pex3, ein integrales Membranprotein, in Hefe essentiell.[6] Der Abbau von Peroxysomen wird als Peroxyphagie bezeichnet, in Analogie zu Mitophagie (Abbau von Mitochondrien) und Reticulophagie (Abbau des ER[7]).
Proteintransport
Da Peroxisomen keine Ribosomen enthalten, müssen alle Enzyme im Zytosol synthetisiert und danach ins Peroxisom transportiert werden.[3] Hierbei werden Proteine posttranslational im gefalteten Zustand ins Peroxisom gebracht.[8] Es sind zwei Wege bekannt. Die meisten Proteine benötigen eine C-terminale Signalsequenz, das sogenannte peroxisom targeting signal (Peroxisom-Zielsignal) PTS1. Diese Signalsequenz ist kürzer als die von Proteinen, die ins Mitochondrium oder ins ER gebracht werden sollen; meistens besteht diese nur aus den drei Aminosäuren Serin-Lysin-Leucin (SKL). Die Signalsequenz jener „PTS1-Proteine“ wird im Cytosol von Pex5p erkannt und zum Peroxisom geführt, wo diese durch einen Proteinmembrankomplex ins Innere des Peroxisoms transportiert werden. Dabei dockt der Protein-Pex5p-Komplex an das integrale Membranprotein Pex14 an.[9] Der Komplex aus Pex5 und dem Protein wird dann in das Peroxisom transportiert, wo Pex5 abgespalten wird und unter Verbrauch von ATP über den Pex2/10/12 Membrankomplex wieder recycelt wird.[10]
Beim zweiten Transportweg wird ein N-terminales und auch längeres Signalpeptid durch Pex7p zum Proteinmembrankomplex des Peroxisoms gebracht. Diese Signalsequenz wird auch als PTS2 bezeichnet, transportierte Proteine sind infolgedessen PTS2-Proteine. Neben Pex7p wird in Säugetierzellen auch eine gespleißte Form von Pex5p verwendet. Nach Transport in die Matrix des Peroxisoms wird dann das Signalpeptid abgeschnitten.
Erkrankungen
Erkrankungen, bei denen Peroxisomen eine Rolle spielen (Peroxisomopathien, Peroxisomale Krankheiten):
1. Peroxisomendefekte
- Zellweger-Syndrom
- Rhizomele Chondrodysplasia punctata Typ 1 (Mutation des PEX7-Gens)
- Neonatale Adrenoleukodystrophie
- Infantiles Refsum-Syndrom
2. Peroxisomaler Enzymdefekt
- Pseudo-Zellweger-Syndrom (Mutation der Acyl-CoA-Oxidase)
- X-chromosomale Adrenoleukodystrophie (sek. durch peroxisomalen Transporterproteindefekt für VLCFA-CoA-Synthetase)
- Rhizomele Chrondrodysplasia punctata Typ 2 (Mutation vom DHAPAT/GNPAT-Gen)
Siehe auch
Literatur
- B. Alberts et al.: Molecular Biology of the Cell. Garland Science, 4. Auflage, 2002. ISBN 0-8153-4072-9.
- N. Campbell et al.: Biologie. 1. Aufl., 1. korrigierter Nachdr., Spektrum Akademischer Verlag 1997, Heidelberg. ISBN 3-8274-0032-5.
Einzelnachweise
- ↑ David Nelson, Michael Cox: Lehninger Biochemie. 4. Auflage. Springer, Berlin / Heidelberg 2009, ISBN 978-3-540-68637-8, S. 876.
- ↑ Soliman K, et al.: Super-resolution imaging reveals the sub-diffraction phenotype of Zellweger
Syndrome ghosts and wild-type peroxisomes. Sci Rep. 2018, 17,7809,
 PMID 29773809
PMID 29773809
- ↑ Hochspringen nach: a b c Peter H. Raven, Ray F. Evert, Susan E. Eichhorn: Biologie der Pflanzen. 4. Auflage. Gruyter, Berlin, New York 2006; ISBN 978-3-11-018531-7; S. 53f.
- ↑ Horst Feldmann: Yeast: Molecular and Cell Biology. Wiley-VCH Verlag GmbH & Co. KGaA 2009; ISBN 978-3-527-32609-9; S. 159.
- ↑ D’Eustachio / reactome: Peroxisomal lipid metabolism
- ↑ Margit Pavelka (Hrsg.) und Jürgen Roth (Hrsg.): Functional Ultrastructure: Atlas of Tissue Biology and Pathology. Springer, Wien; 2. Auflage 2010; ISBN 978-3-211-99389-7; S. 134.
- ↑ Daniel J. Klionsky et al. (2007): How shall I eat thee? In: Autophagy 3(5); S. 413–416;
PMID 17568180;
PDF (freier Volltextzugriff, englisch).
- ↑ Lynne Cassimeris, George Plopper und Vishwanath R. Lingappa: Lewin's Cells. Jones & Bartlett Pub (Ma); 2. Auflage 2010; ISBN 978-0-7637-6664-1; S. 338
- ↑ Marc Fransen, Stanley R. Terlecky, and Suresh Subramani: Identification of a human PTS1 receptor docking protein
directly required for peroxisomal protein import,
PMC 20933 (freier Volltext)
- ↑ Harvey Lodish: Molecular Cell Biology (Seventh Edition, 2012) S. 612f. ISBN 978-1-4641-0981-2


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 27.06. 2026