Hydroformylierung
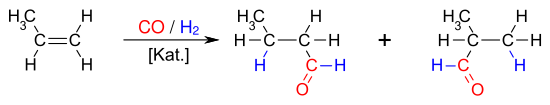
Die Hydroformylierung (auch: Oxosynthese, seltener Roelen-Synthese oder Roelen-Reaktion) ist eine technisch bedeutende, homogen katalysierte Reaktion von Olefinen mit Kohlenstoffmonoxid und Wasserstoff. Die primären Produkte der Hydroformylierung sind Aldehyde mit einem Kohlenstoffatom mehr als das Olefinsubstrat. Diese Aldehyde werden zur Herstellung einer Vielzahl nützlicher Folgeprodukte verwendet, die Aldehyde selbst haben eine geringe Verwendbarkeit. Wichtige industrielle Produkte der Hydroformylierung sind 1-Butanol und 2-Ethylhexanol, die beide aus Propen gewonnen werden. Die Produkte der Hydroformylierung werden vielfältig als Lösungsmittel oder als Zwischenprodukte für die Herstellung von Wasch- und Reinigungsmitteln, Schmiermitteln oder Weichmachern für Kunststoffe eingesetzt. Im Jahr 2008 betrug die mittels Hydroformylierung hergestellte Menge an Oxo-Produkten etwa 10,4 Millionen Tonnen.
Die Entdeckung dieser metallorganischen Komplexkatalyse gelang dem deutschen Chemiker Otto Roelen im Dezember 1937 bei der Untersuchung der Fischer-Tropsch-Synthese in den Forschungslaboratorien der Ruhrchemie. Das Verfahren gilt als eine der bedeutendsten Entwicklungen der industriellen Chemie des 20. Jahrhunderts und als erster großtechnischer homogenkatalytischer Prozess. Als Hydroformylierungs-Katalysatoren verwendet die chemische Industrie metallorganische Cobalt- oder Rhodiumverbindungen. Das industrielle Verfahren wird bei Drücken von etwa 10 bar bis 100 bar und Temperaturen zwischen 40 und 200 °C durchgeführt. Die Kapazität der industriellen Anlagen wurde seit seiner Erfindung kontinuierlich ausgebaut, die Gesamtkapazität der Hydroformylierungsanlagen beträgt mehrere Millionen Tonnen pro Jahr, wobei die Produkte auf Propenbasis den größten Volumenanteil haben. Die Hydroformylierung nach dem zweiphasigen Ruhrchemie/Rhône‐Poulenc‐Verfahren auf Basis von wasserlöslichen Rhodiumkatalysatoren war einer der ersten technisch wichtigen homogen-katalysierten Prozesse, bei denen das entscheidende Problem der Rückführung des Katalysators zufriedenstellend gelöst wurde.
Neben den industriellen Anwendungsmöglichkeiten stellt die Hydroformylierung eine ideale atomökonomische Reaktion zur Bildung von Kohlenstoff-Kohlenstoff-Bindungen mit einzigartigen Möglichkeiten zur Anwendung in der zielgerichteten organischen Synthese dar. Durch den Einsatz von Katalysatorsystemen mit maßgeschneiderten Liganden, welche die Regio-, Stereo- und Enantioselektivität steuern, wurde die Hydroformylierung ein wichtiges Werkzeug in der organischen Synthese von Feinchemikalien.
Geschichte
O. C. Elvins und A. W. Nash wiesen bei ihren Untersuchungen zur Fischer-Tropsch-Synthese 1926 erstmals auf die Bildung von sauerstoffhaltigen Komponenten bei dieser Reaktion hin. Auch D. F. Smith, C. O. Hawk und P. L. Golden beobachteten die Bildung von sauerstoffhaltigen Verbindungen bei der Reaktion von Ethylen mit Kohlenstoffmonoxid und Wasserstoff unter Fischer-Tropsch-Bedingungen. Sie erhielten eine Mischung von Aldehyden und Alkoholen, wobei ein Großteil des eingesetzten Ethens zu Ethan hydriert wurde. Ihre Versuche, die Prozessführung in Hinblick auf sauerstoffhaltige Verbindungen zu optimieren, gelangen aber nicht.
Entdeckung

Im Werk der Ruhrchemie in Oberhausen entdeckte Otto Roelen die Hydroformylierung im Dezember 1937 zufällig beim Versuch, das bei der Fischer-Tropsch-Synthese anfallende Ethen in den Prozess zurückzuführen. Bei Versuchen, in denen neben Ethen auch Ammoniak der Fischer-Tropsch-Synthese zugeführt wurde, fand Roelen Ablagerungen von Propionaldimin, einem Kondensationsprodukt aus Ammoniak und Propionaldehyd. Im Gegensatz zu anderen Forschern interpretierte er die Bildung von Propionaldehyd als eigenständige Reaktion, die er auf die Zugabe von Ethen zurückführte und nicht als Nebenreaktion der Fischer-Tropsch-Synthese ansah.
Nach ersten Versuchen zur Optimierung der Reaktion in Richtung der Aldehydbildung, die er im Juli 1938 begann, reichte Roelen bereits Ende desselben Jahres ein Patent für die Oxosynthese ein. Der Name „Oxosynthese“ beruht auf der falschen Vermutung, dass es sich um eine generelle Synthese zur Herstellung von Oxo-Produkten wie Aldehyden und Ketonen handelte. Ketone fallen aber nur bei der Hydroformylierung von Ethen als Folgeprodukt in Form von Diethylketon in großen Mengen an. Die Ruhrchemie, Otto Roelens Arbeitgeber, wählte als Lösungsmittel für die Hydroformylierung Toluol, da es sich leicht von den entstehenden Produkten abtrennen ließ. Bei einer Reaktionstemperatur von 115 °C erzielte der Prozess bei vollständigem Umsatz des Ethens eine Ausbeute von 70 bis 80 % Propionaldehyd, 15 % organische Nebenprodukte und 5 % Verlust.
Technischer Prozess
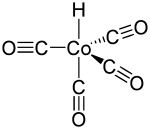
Als Katalysator verwendete Roelen einen Cobaltoxid, Thorium(IV)-oxid und Magnesiumoxid enthaltenden Katalysator, der sonst für die Fischer-Tropsch-Synthese eingesetzt wurde. Er fand jedoch, dass viele andere Cobaltsalze als Katalysator-Precursor geeignet waren und vermutete, dass Cobaltcarbonylhydrid die aktive Katalysatorspezies war. Dieser Komplex gilt als erste Katalysatorgeneration.
Die I. G. Farben verwendete eine Prozessvariante mit einem auf Bimsstein fixierten Katalysator in einem wässrigen System. Als Einsatzstoffe nutzte die I. G. Farben ein äquimolares Gemisch von Ethen, Kohlenstoffmonoxid und Wasserstoff bei Reaktionstemperaturen von 150 bis 200 °C und einem Reaktionsdruck von 15 bis 30 MPa (150 bis 300 bar). Bei dieser Prozessvariante wurde etwa 20 bis 25 % des Ethens zum Ethan hydriert, die Selektivität zum Propionaldehyd betrug nur 65 %. Der Verlust an Katalysator wurde durch Zugabe von Cobaltfettsäuresalzen ausgeglichen.
Roelen arbeitete bereits früh an der Hydroformylierung von Fischer-Tropsch-Olefinen mit einer Kettenlänge von 11 bis 17 Kohlenstoffatomen für die Herstellung von Fettalkoholen. Im Jahr 1940 begann die Ruhrchemie mit dem Bau einer Anlage, deren Kapazität 7000 Jahrestonnen Fettalkohole betragen sollte. Die Ruhrchemie nahm die Anlage im Krieg aber nicht mehr in Betrieb.
Weitere Entwicklungen
Aufgrund der niedrigen Selektivität zum n-Isomeren entwickelte die Shell Katalysatorsysteme, bei denen der Kohlenstoffmonoxidligand des Cobaltcarbonylhydrids teilweise durch Phosphane ersetzt war. Durch Ligandenmodifikation war es nun möglich, die Selektivität zum gewünschten Produkt zum Teil zu steuern. Die Komplexe des Typs HCo(CO)3(PR3) gelten als zweite Katalysatorgeneration, die Prozesstemperaturen von 150 bis 190 °C und Drücke von 40 bis 80 bar bei einer n/iso-Selektivität von 88:12 erlaubten.
Lauri Vaska berichtete im Jahr 1963 über die Darstellung des Komplexes Rhodiumtetracarbonylhydrid. Diesen Komplex und seine Triphenylphosphananaloga setzte Geoffrey Wilkinson ab 1968 für die Hydroformylierung ein. Dies gilt als Meilenstein in der Weiterentwicklung des Verfahrens, da Rhodiumcarbonylhydridkomplexe mit Triphenylphosphan-Liganden eine hohe Aktivität und Selektivität in der Hydroformylierung aufweisen. Die Firma Union Carbide nutzte ab 1976 Katalysatoren des Typs HRh(CO)(PR3)3 für die Entwicklung des Low-Pressure-Oxo-Prozesses (LPO), der bei Temperaturen von 90 bis 100 °C und Drücken von 15 bis 18 bar arbeitet. Durch die geringe Hydrierungsaktivität des Katalysators, mildere Reaktionsbedingungen sowie eine hohe n/iso-Selektivität erlangte das Verfahren trotz des höheren Rhodiumpreises schnell technische Bedeutung. Phospanmodifizierte Rhodiumcarbonylhydride gelten als dritte Generation der Hydroformylierungskatalysatoren.
Neben dem ursprünglichen Verfahren entwickelten weitere Firmen wie BASF und Shell Verfahrensvarianten, die bei Drücken von etwa 5 MPa (50 bar) und Temperaturen von etwa 100 °C arbeiten. Die Prozesse beruhen alle auf phosphanmodifizierten Cobalt- und Rhodiumkomplexen. Shell nutzte ein tributylphosphanmodifiziertes Cobaltcarbonylhydrid zur Hydroformylierung längerkettiger Olefine, die direkt zum Alkohol weiterhydriert wurden.
Zwei-Phasen-Hydroformylierung

Es wurden zahlreiche Untersuchungen zur Fixierung des Katalysators unternommen, mit dem Ziel, die Trennung von Produkt- und Katalysatorphase zu erleichtern. Es wurde etwa versucht, durch die Kombination von Diphenylphosphinomethylpolystyrol und Dicobaltoctacarbonyl eine Fixierung zu erreichen. Diese Bemühungen hatten jedoch nicht den gewünschten Erfolg. Probleme mit der Deaktivierung des Katalysators und seines Austrags verbesserten sich in den 1980er Jahren entscheidend durch die Einführung wasserlöslicher Katalysatoren im Ruhrchemie/Rhône-Poulenc-Verfahren. Basierend auf dem von Wilkinson entwickelten Rhodiumtristriphenylphosphancarbonylhydrid und den Arbeiten von Kuntz bei Rhône Poulenc entwickelte die Ruhrchemie innerhalb von 24 Monaten und mit einem Scale-Up-Faktor von 1 : 24.000 den technischen Prozess. Der Katalysator verbleibt dabei in der Wasserphase, die leichtere, in Wasser unlösliche Produktphase kann dadurch leicht vom Katalysator getrennt werden. Die wasserlöslichen Rhodiumkatalysatoren gelten als vierte Generation der Hydroformylierungskatalysatoren.
Seit Mitte der 1990er Jahre wird versucht, durch Einsatz von Lösungsmitteln wie etwa überkritisches Kohlenstoffdioxid. perfluorierte Systeme oder ionische Flüssigkeiten die Reaktion weiter zu optimieren. Des Weiteren wurden neben dem Ruhrchemie/Rhône-Poulenc-Verfahren andere Wege und Methoden der Heterogenisierung erprobt. Dabei konnte meist das Problem des Katalysatoraustrags nicht gelöst werden. Am 24. August 2013, zum 75. Jubiläum der Patenteinreichung, wurde das Werk Ruhrchemie von der GDCh in das Programm Historische Stätten der Chemie aufgenommen.
Rohstoffe
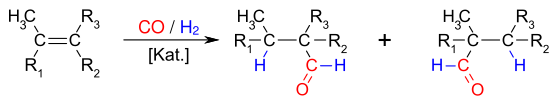
Viele Olefine (Alkene und Cycloalkene) sind der Hydroformylierung zugänglich. Es handelt sich formal um eine Addition von Wasserstoff und einer Formylgruppe an die Doppelbindung eines Olefins, wobei die Kombination Olefin/Katalysator eine wesentliche Rolle für die zu erzielenden Umsätze und Selektivitäten spielt.
Synthesegas
Die Herstellung von Synthesegas kann prinzipiell aus festen, flüssigen oder gasförmigen Ausgangsstoffen erfolgen. Als Kohlenstoffquelle können sowohl Kohle (im Formelbeispiel als C) über die Kohlevergasung, Erdöl (C5H12), Erdgas (CH4) als auch nachwachsende Rohstoffe über Dampfreformierung oder Partielle Oxidation dienen.



Die Ruhrchemie nahm 1986 eine Kohlevergasung in der Synthesegasanlage Ruhr nach dem Prinzip eines Texaco-Kohlevergasers in Betrieb. Dabei wurde eine Feinkohle/Wasser-Slurry bei Temperaturen von etwa 1200 °C und einem Druck von etwa 20 bar zu Synthesegas umgesetzt. Der Durchsatz betrug 30 Tonnen Steinkohle pro Stunde, wodurch 40.000 Normkubikmeter Synthesegas sowie 10.000 Normkubikmeter Wasserstoff entstanden.
In der Praxis ist ein Verhältnis von Kohlenstoffmonoxid zu Wasserstoff von etwa 40 : 60 erwünscht. Der überschüssige Wasserstoff wird in der parallel ablaufenden Hydrierung der Aldehyde zu Alkoholen verbraucht.
Olefine
Kurzkettige Olefine reagieren meist schneller als längerkettige Olefine und Cycloolefine, lineare Olefine reagieren schneller als verzweigte. Fast alle Olefine lassen sich hydroformylieren, wobei die Reaktion von vierfach mit Alkylgruppen substituierten Olefinen, die zu einem quaternären Kohlenstoffatom führen würden, nur selten gelingt. Styrol lässt sich durch Cobaltkatalysatoren kaum hydroformylieren, mit Rhodiumkatalysatoren werden dagegen hohe Umsätze erzielt.
Propen, eine ungesättigte organische Verbindung mit der chemischen Formel C3H6, ist der bei weitem häufigste Rohstoff für die Hydroformylierung. Propen wird aus fossilen Rohstoffen wie Erdöl oder Erdgas und im geringeren Umfang aus Kohle gewonnen. Propen fällt beim Cracken von Naphtha und als Nebenprodukt der Erdgasverarbeitung an. Eine weitere wichtige petrochemische Propenquelle ist die Dehydrierung von Propan.
Konjugierte Diene
Konjugierte Diene lassen sich mit Rhodium-Phosphan-Katalysatoren zum Dialdehyd hydroformylieren, Cobaltkatalysatoren liefern durch Hydrierung einer Doppelbindung überwiegend Monoaldehyde. Nichtkonjugierte Diene lassen sich zu Dialdehyden hydroformylieren, wenn die Doppelbindungen in der Kette mindestens durch zwei Kohlenstoff-Kohlenstoff-Einfachbindungen getrennt sind. Allylalkohole, Allylester und Allylether lassen sich bevorzugt mit isomerisierungsfreien Katalysatoren hydroformylieren. α,β-ungesättigte Ketoverbindungen reagieren meist unter Hydrierung der Doppelbindung. Ungesättigte Carbonsäuren und Carbonsäureester lassen sich gut hydroformylieren, ebenso ungesättigte Aldehyde und Ketone mit nichtkonjugierten Doppelbindungen.
Funktionalisierte Olefine
Neben reinen Olefinen können funktionalisierte Olefine wie Allylalkohol hydroformyliert werden. Als Zielprodukt wird mit isomerisierungsfreien Katalysatoren wie Rhodium-Triphenylphosphan-Komplexen 1,4-Butandiol und sein Isomeres erhalten. Beim Einsatz des Cobaltkomplexes wird durch Isomerisierung der Doppelbindung n-Propanal erhalten. Die Hydroformylierung von Alkenylethern und Alkenylestern erfolgt meist in α-Position zur Ether- oder Esterfunktion. Der hydroformylierte Ester kann durch nachfolgende Abspaltung der Carbonsäure aus dem Hydroformylierungsprodukt zu α,β-ungesättigten Aldehyden führen.
Die Hydroformylierung von Acrylsäure und Methacrylsäure untersuchte Jürgen Falbe. Danach bildet sich bei der rhodium-katalysierten Variante im ersten Schritt das Markownikoff-Produkt. Durch die Wahl der Reaktionsbedingungen lässt sich die Reaktion in verschiedene Richtungen lenken. Eine hohe Reaktionstemperatur und niedrige Kohlenstoffmonoxiddrücke begünstigen die Isomerisierung des Markownikoff-Produkts zum thermodynamisch stabileren β-Isomer, das zum n-Aldehyd führt. Niedrige Temperaturen, hohe Kohlenstoffmonoxiddrücke und ein Überschuss von Phosphinen, die freie Koordinationsstellen besetzen können, führen zur schnelleren Hydroformylierung in α-Position zur Estergruppe und unterdrücken die Isomerisierung.
Die Hydroformylierung von konjugierten Olefinen führt mit vielen Katalysatorsystemen durch Hydrierung einer Doppelbindung zu denselben Produkten wie die entsprechenden Monoolefine. Mit Rhodium-Phosphin-Komplexen führt die Hydroformylierung zu Dialdehyden. Die Hydroformylierung von Alkinen führt zu α,β-ungesättigten Aldehyden.
Katalysatoren
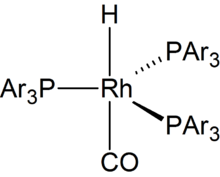
Als Hydroformylierungskatalysatoren werden Metallcarbonylhydride und deren Derivate eingesetzt. Sie können durch die allgemeine Formel

beschrieben werden. Die verwendeten Katalysatoren haben eine eindeutig definierte Struktur und lassen sich genau charakterisieren und in gleich bleibender Qualität synthetisieren oder in-situ generieren. Im katalytischen Prozess sind alle Metallatome als aktive Zentren für die Synthese des Produktes zugänglich.
Die chemische Industrie verwendet Cobalt- und Rhodiumkomplexe mit verschiedenen Kohlenstoffmonoxid-, Phosphin- und Phosphit-Liganden. Komplexe mit Metallen der Eisengruppe wie Eisen, Ruthenium und Osmium, aber auch Iridium sowie polymetallische Systeme wie Platin/Zinn wurden untersucht, wiesen jedoch nicht die gleiche Aktivität wie Rhodium und Cobaltkatalysatoren auf. Rhodiumkomplexe sind die aktivsten Hydroformylierungskatalysatoren und etwa 1000-mal aktiver als Cobaltkomplexe. Die Hydroformylierungsaktivität des metallorganischen Katalysatorkomplexes sinkt vom Cobaltkomplex zu den Iridium-, Ruthenium-, Osmium-, Mangan- und Eisenkomplexen jedes Mal etwa um den Faktor 10. Auch metallisches Calcium ist als Hydroformylierungskatalysatoren wirksam, weniger aktiv sind Magnesium und Zink. Es wird vermutet, dass die Aktivität der Metalle sich auf die Bildung von Hydriden zurückführen lässt. Für industrielle Anwendungen sind lediglich Rhodium- und Cobaltkomplexe von Bedeutung.
Cobaltkatalysatoren
Für die ersten Hydroformylierungen wurde der Katalysator der Fischer-Tropsch-Synthese verwendet, der aus etwa 30 % Cobalt, 2 % Thoriumoxid, 2 % Magnesiumoxid und 66 % Kieselgur bestand. Dieser scheinbar heterogene Katalysator wurde zusammen mit den organischen Reaktanden aufgeschlämmt. Diese Aufschlämmung, auch Maische genannt, wurde dann in eine Hochdruckkammer gepumpt, wo unter Hochdruck die Hydroformylierung erfolgte. Nachdem die Reaktion beendet war, wurde der nun gelöste Cobaltkatalysator durch Wasserstoff reduziert und auf dem Kieselgur adsorbiert. Später wurden als Katalysatorvorstufen öllösliche Cobaltsalze wie Naphthenate eingesetzt.
Unter den Reaktionsbedingungen bildet sich aus den Cobaltsalzen oder feinverteilten Cobaltmetall zunächst Dicobaltoctacarbonyl. Durch Aufnahme von Wasserstoff bildet sich daraus der von Walter Hieber entdeckte einkernige Komplex Cobaltcarbonylhydrid (CoH(CO)4), der eigentliche Katalysator der Hydroformylierung.
![{\displaystyle \mathrm {[Co_{2}(CO)_{8}]\ +\ H_{2}\longrightarrow \ 2\ [CoH(CO)_{4}]} }](/svg/a4f80426e35289df4aa70d29aa181a9541c9ca69.svg)
Liganden
Das Ligandendesign hat einen großen Einfluss auf das n-/iso-Verhältnis der entstehenden Produkte, vor allem durch sterische Effekte. Weiterhin hat das Verhältnis von Metall zu Ligand einen Einfluss auf die n-/iso-Selektivität und die Nebenreaktionen. Geringe Zusätze tertiärer Amine können die Reaktion beschleunigen; höhere Konzentrationen können dagegen zur vollständigen Unterdrückung der Reaktion führen. Beim Einsatz von Rhodium als Katalysatormetall ist die katalytisch aktive Spezies ein trigonal-bipyramidaler Rhodiumcarbonylhydrido-Komplex, der in zwei isomeren Formen vorliegt. Die zwei Phosphanliganden besetzen entweder eine äquatorial-äquatoriale (ee) oder die äquatorial-apicale (ea) Position. Durch Einsatz von bidentaten Diphosphanliganden wurden gute Selektivitäten zum linearen Aldehyd gefunden. Der Einfluss des sogenannten Bisswinkels der Diphosphanliganden wurde eingehend untersucht.
Neben den sterischen Effekten beeinflussen die elektronischen Effekte des Liganden die Katalysatoraktivität. Gute π-Akzeptoren wie Phosphite senken in Rhodiumkomplexen durch eine starke Rückbindung die Elektronendichte am Metall und schwächen dementsprechend die Rhodium-Kohlenstoffmonoxid-Bindung. Die Insertion des Kohlenstoffmonoxids in die Metall-Alkyl-Bindung wird dadurch erleichtert. Metallorganische Rhodiumphosphitkomplexe sind daher sehr gute Hydroformylierungskatalysatoren.
Reaktionstechnik
Die technischen Verfahren unterscheiden sich nach der Kettenlänge des zu hydroformylierenden Olefins in der Art des Katalysatormetalls sowie der Abtrennung des Katalysators. Das ursprüngliche Verfahren der Ruhrchemie setzte Ethen mittels Cobaltcarbonylhydrid zu Propanal um. Heute werden Prozesse mit auf Cobalt basierenden Katalysatoren hauptsächlich für die Produktion von mittel- bis langkettigen Olefinen eingesetzt, während auf Rhodium basierende Katalysatoren meist für die Hydroformylierung von Propen verwendet werden. Die Rhodiumkatalysatoren sind wesentlich teurer als Cobaltkatalysatoren. Bei der Hydroformylierung höhermolekularer Olefine ist die verlustfreie Abtrennung vom Katalysator schwierig. Die Verfahren unterscheiden sich hauptsächlich in der Art der Katalysatorabtrennung und der Rückgewinnung des Katalysators.
Produkte

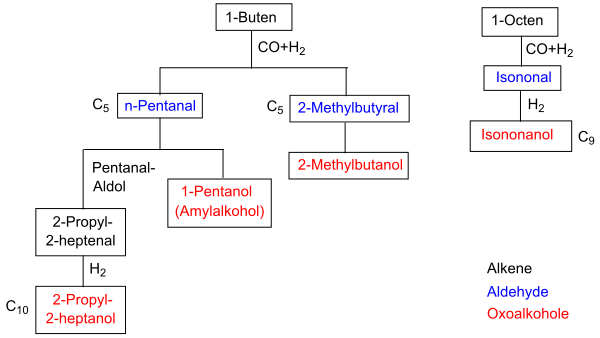
Die primären Produkte der Hydroformylierung sind Aldehyde, die durch Aldolkondensation, Hydrierung, Oxidation, Aminierung und andere Verfahren zu einer breiten Vielfalt von Folgeprodukten weiterverarbeitet werden. Über 70 % der industriellen Gesamtproduktion entfällt auf die Produktion von n-Butanal beziehungsweise n-Butanol, etwa 20 % auf die Produktion von C5- bis C13-Aldehyden, der Rest entfällt auf höhermolekulare Aldehyde und Propanal.
Die Weiterverarbeitung der primär entstehenden Aldehyde kann in einer zweistufigen Sequenz erfolgen, bei der die Hydrierung direkt auf die Hydroformylierung in einem separaten oder im gleichen Reaktionsgefäß erfolgt oder durch eine Hydroformylierung unter reduzierenden Bedingungen. Des Weiteren ist eine mehrstufige Weiterverarbeitung möglich. Dabei reagieren die Aldehyde zunächst in einer Reaktion wie der Aldolkondensation zu Folgeprodukten, bevor die Hydrierung zum Alkohol erfolgt.
n-Butanol
Das bei der Hydroformylierung von Propen primär entstehende Butanal reagiert mit Wasserstoff weiter zu n-Butanol. n-Butanol ist ein bedeutendes Zwischenprodukt zur Herstellung von Butylestern wie Butylacrylat, Butylacetat und Dibutylphthalat. n-Butanol wird weiterhin zur Herstellung von Arzneimitteln, Polymeren, Herbiziden, Katalysatoren und vielen weiteren Anwendungen verwendet. Sowohl n-Butanol und als auch Isobutanol können rein oder als Beimischung zu Kraftstoffen für Ottomotoren verwendet werden. Diese Butanole weisen im Vergleich zu Ethanol eine höhere Energiedichte auf.
2-Ethylhexanol
Das bei der Hydroformylierung von Propen primär anfallende n-Butanal reagiert unter basischer Katalyse zu 2-Ethylhexenal, welches durch Dehydratisierung und anschließende Hydrierung zu 2-Ethylhexanol (2-EH) umgesetzt wird.
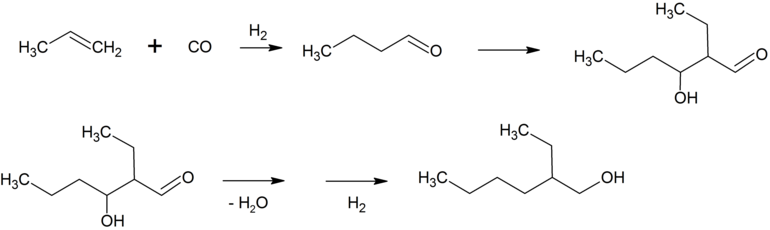
Dieser Alkohol wird mit Phthalsäureanhydrid zu Bis(2-ethylhexyl)phthalat, das allgemein vereinfachend als Dioctylphthalat (DOP) bezeichnet wird, umgesetzt. Bis(2-ethylhexyl)phthalat ist ein wichtiger Weichmacher für Polyvinylchlorid (PVC). Etwa 2,5 Millionen Tonnen Bis(2-ethylhexyl)phthalat werden jährlich produziert. Etwa 25 % des n-Butanals wird zu n-Butanol hydriert, das als Lösungsmittel und für Veresterungen verwendet wird. Die höhermolekularen Aldehyde, die zum Beispiel durch Hydroformylierung von SHOP-Olefinen mit Cobaltkatalysatoren erhalten werden, werden meist zu Fettalkoholen hydriert. Diese werden, oft nach Ethoxylierung, sulfatiert und nach Neutralisation mit Natronlauge oder Ammoniak als anionische Tenside verwendet.
Laborsynthesen
Prochirale Olefine lassen sich mit chiralen Komplexen enantioselektiv hydroformylieren. So lässt sich beispielsweise Dexibuprofen, das (+)-(S)-Enantiomer des Ibuprofen, durch enantioselektive Hydroformylierung und anschließende Oxidation herstellen. Durch Einsatz chiraler Phosphanliganden wie DIOP, DIPAMP oder BINAPHOS lassen sich hohe Enantiomerenüberschuss erzielen. Die asymmetrische Hydroformylierung dient unter anderem der Synthese chiraler Produkte, die in der Pharmazie oder als Aktivkomponente für Agrochemikalien eingesetzt werden. Im Gegensatz zur technischen Hydroformylierung ist das Zielprodukt der verzweigte Aldehyd mit einem Chiralitätszentrum.
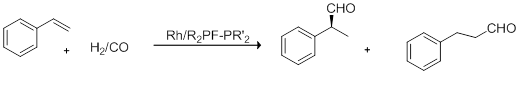
Nur das Produkt der Markownikow-Addition führt zum Zielprodukt, während der n-Aldehyd achiral ist. Die Stereochemie wird im Schritt der Olefinkoordination festgelegt.
Die Hydroformylierung lässt sich durch den Ersatz von Wasserstoff durch Monohydrosilanen (H-Si-R3) zur so genannten Silylformylierung modifizieren. Dabei wird eine Trialkylsilylgruppe und eine Formylgruppe an die Dreifachbindung eines Alkins und Bildung eines 3-Silyl-2-alkenals addiert.
Die Tandem-Reaktion von Hydroformylierung mit zum Beispiel Knoevenagel-Reaktionen, Wittig-Olefinierungen oder Allylborierungen ermöglicht den Aufbau komplexer Moleküle, die sich zum Teil, etwa in Kombination mit einer reduktiven Aminierung als Hydroaminomethylierung, in einer Eintopfreaktion durchführen lassen.
Reaktionsmechanismus
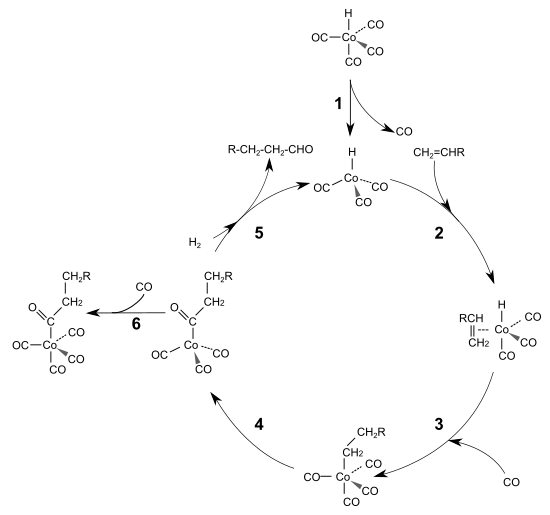
Den sogenannten Heck-Breslow-Mechanismus der cobaltkatalysierten Hydroformylierung klärten 1960 der späteren Nobelpreisträger Richard F. Heck und David Breslow auf. Demnach wird zunächst ein Kohlenstoffmonoxidligand aus Cobaltcarbonylhydrid unter Bildung einer 16-Elektronen-Spezies eliminiert (1). Dies schafft eine freie Koordinationsstelle, an die sich ein Olefin unter Ausbildung einer 18-Elektronen-Spezies mittels π-Bindung anlagern kann (2). Im nächsten Schritt folgt die Bildung eines 16-Elektronen-Alkylkomplexes, in dessen freie Koordinationsstelle ein Kohlenstoffmonoxidligand aufgenommen wird (3). Dieser insertiert in die Metall-Kohlenstoffbindung des Alkylrestes wiederum unter Bildung eines 16-Elektronen-Acylkomplexes (4). Durch oxidative Addition von Wasserstoff wird der Aldehyd freigesetzt und die aktive Spezies wiederhergestellt (5). Unter Bildung des Ausgangskomplexes schließt sich der katalytische Kreislauf. Als Nebenreaktion kann der 16-Elektronen-Komplex in einer Gleichgewichtsreaktion ein Molekül Kohlenstoffmonoxid aufnehmen (6). Sowohl bei der cobalt- als auch bei der rhodiumkatalysierten Hydroformylierung gilt die oxidative Addition des Wasserstoffs als geschwindigkeitsbestimmender Schritt mit anschließender reduktiver Eliminierung des Aldehyds.
Rhodiumhydridocarbonyle und deren phosphanmodifizierte Analoga reagieren nach einem gleichartigen Mechanismus, den im Jahr 1968 Geoffrey Wilkinson untersuchte. Im ersten Schritt dissoziiert demnach ein Phosphanligand aus dem Komplex und bildet eine planare, koordinativ ungesättigte 16-Elektronen-Spezies. An diese koordiniert ein Olefin unter Bildung eines 18-Elektronen-Komplexes. Nach Insertion des Olefins in die Rhodium-Wasserstoff-Bindung unter Alkylkomplexbildung und nach Anlagerung eines weiteren Moleküls Kohlenstoffmonoxid insertiert dieses in die Rhodium-Alkylbindung unter Ausbildung des Acylkomplexes. Unter Anlagerung von Kohlenstoffmonoxid schließt sich der katalytische Zyklus und der Ausgangskomplex wird wiederhergestellt.
Die Ausbildung der quadratisch-planaren Zwischenstufe mit zwei sterisch anspruchsvollen Phosphanliganden gilt als Erklärung für das hohe n-/iso-Verhältnis der rhodiumkatalysierten Hydroformylierung. Die sterischen Zwänge im Übergangszustand bewirken, dass der Alkylligand bevorzugt linear koordiniert wird. Dies erklärt, warum das n-/iso-Verhältnis positiv durch zunehmende Phosphankonzentration und sinkenden Kohlenstoffmonoxid-Partialdruck beeinflusst wird.
Der Mechanismus wurde mittels infrarotspektroskopischen und Hochdruck-NMR-Methoden untersucht. Die Verwendung von Deuterium in der Reaktion (Deuteroformylierung) erlaubt die Untersuchung der entstehenden Produkte mittels 1H-NMR-Spektroskopie und damit Rückschlüsse auf den Mechanismus.
Regioselektivität
Ein wichtiges Kriterium der Hydroformylierung ist die Regioselektivität zum n- oder iso-Produkt. So wird das n-Isomer der Propenhydroformylierung im industriellen Maßstab zu 2-Ethylhexanol weiterverarbeitet, wohingegen das iso-Isomer nur untergeordnete Bedeutung für die Darstellung von Isobutanol, Neopentylglycol oder iso-Buttersäure hat. Der Schritt der Insertion des Olefins in die Metall-Wasserstoffbindung unter Bildung des Alkylkomplexes entscheidet mit über die Selektivität der Bildung von n- oder iso-Aldehyden:
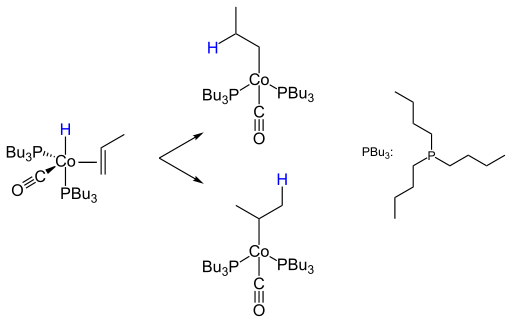
Bei höhermolekularen Olefinen kann es zu einer Isomerisierung der Doppelbindung kommen, wodurch Gemische von Aldehyden gebildet werden. In die Metall-Kohlenstoff-Bindung des Alkylkomplexes insertiert ein Kohlenstoffmonoxidligand unter Bildung eines Acylkomplexes.
Keulemans stellte 1948 Regeln für die Produktverteilung auf (Keulemans-Regeln) Demnach wird aus geradkettigen Olefinen stets ein Gemisch von n- und 2-Alkylalkoholen im Verhältnis von 40 bis 60 % n- und 60 bis 40 % 2-Alkylalkoholen. Eine Addition der Formylgruppe an tertiäre Kohlenstoffatome findet nicht statt; iso-Buten bildet beispielsweise nur das 3-Methylbutanol. Die Anlagerung an Kohlenstoffatome in α-Stellung zu tertiären Kohlenstoffatomen ist sterisch gehindert, kann jedoch stattfinden. Zur Anlagerung an Kohlenstoffatome in α-Stellung zu quartären Kohlenstoffatomen kommt es nicht. Ein isoliertes tertiäres Kohlenstoffatom behindert die Bildung der möglichen Isomere. Die Hydroformylierung wird immer von einer Doppelbindungsisomerisierung begleitet. Außer der 2-Alkylverzweigung besteht keine Tendenz zur Erhöhung des Verzweigungsgrades. Bevorzugt wird unter Verschiebung der Doppelbindung ein lineares Produkt erhalten.
Kinetische Untersuchungen lieferten für die cobaltkatalysierte Reaktion die folgende Geschwindigkeitsgleichung:

Die Reaktion ist mit etwa 125 kJ mol−1 exotherm. Bei rhodiumkatalysierten Hydroformylierungen werden Wechselzahlen von etwa 6000 molOlefin molKatalysator−1 h−1 erreicht.
Neben- und Folgereaktionen
Reaktionen der Olefine
Nebenreaktionen der Olefine sind die Isomerisierung und Hydrierung der olefinischen Doppelbindung. Während die durch Hydrierung der Doppelbindung entstehenden Alkane nicht weiter an der Reaktion teilnehmen, ist die Isomerisierung der Doppelbindung unter späterer Bildung der n-Alkylkomplexe ein gewünschter Vorgang. Die Hydrierung ist meist von untergeordneter Bedeutung. Cobaltphosphan-modifizierte Katalysatoren können jedoch eine erhöhte Hydrieraktivität aufweisen, wobei bis zu 15 % des Olefins hydriert werden.
Reaktionen der Aldehyde
Eine meist gewünschte Nebenreaktion ist die Hydrierung der Aldehyde zu Alkoholen. Höhere Temperaturen und Wasserstoffpartialdrücke begünstigen die Hydrierung der entstehenden Aldehyde zum Alkohol. Die Kinetik der Alkoholbildung mit Cobaltkomplexen lässt sich mit folgender Gleichung beschreiben:

Als Reaktionsmechanismus wird angenommen, dass sich zunächst der π-Komplex des Aldehyds mit dem Katalysator bildet. Unter Umlagerung zum Alkoholat und anschließender oxidativer Addition von Wasserstoff werden der Alkohol und der Ausgangskomplex gebildet:



Die aldehydische Kohlenstoff-Sauerstoff-Doppelbindung kann ebenfalls der Hydroformylierung unterliegen und zu Ameisensäure und ihren Estern führen. Dabei wird in die Sauerstoff-Metall-Bindung Kohlenmonoxid insertiert. Der entstehende Formylkomplex kann unter oxidativer Addition von Wasserstoff den Ameisensäureester freisetzen:


Die primär gebildeten Aldehyde können ebenfalls weiterreagieren und durch Aldolkondensation Produkte wie die Zielproduktvorstufe 2-Ethylhexenal oder höhermolekulare Kondensationsprodukte, sogenanntes Dicköl, bilden.
Homologierung der Alkohole
Unter den Reaktionsbedingungen der Hydroformylierung können die entstehenden Alkohole durch einen sauren Katalysator wie Cobaltcarbonylhydrid wieder zum Olefin dehydratisiert werden. Durch eine weitere Hydroformylierung des Olefins kommt es zu einer Kettenverlängerung und Bildung eines Aldehyds. Dieser wird wieder zum Alkohol reduziert, der wiederum Olefin dehydratisiert werden kann. Die Reaktionsbereitschaft ist bei tertiären Alkoholen am höchsten, sekundäre Alkohole reagieren langsamer und primäre Alkohole reagieren träge. Aus tert-Butanol entsteht etwa Isovaleraldehyd. Selbst Methanol und Benzylalkohol lassen sich zu Ethanol beziehungsweise 2-Phenylethanol homologieren.
Reaktionen des Katalysatorkomplexes
Die eingesetzten Triphenylphosphinkomplexe können unter Reaktionsbedingungen durch Hydrierung Benzol freisetzen. Die Insertion von Kohlenstoffmonoxid in eine intermediäre Metall-Kohlenstoff-Bindung kann zur Bildung von Benzaldehyd oder durch nachfolgende Hydrierung zu Benzylalkohol führen. Der Ligand kann Propen anlagern, wobei das entstehende Diphenylpropylphosphin auf Grund seiner erhöhten Basizität die Reaktion inhibieren kann.
Spurenverunreinigungen der Edukte mit Sauerstoff oder Schwefel und deren Verbindungen können zur Oxidation von Phosphor(III)- zu Phosphor(V)-Verbindungen führen beziehungsweise zu katalytisch inaktiven Metalloxiden und -sulfiden.
Verfahrensvarianten
Ruhrchemie/Rhône-Poulenc-Verfahren

Im Ruhrchemie/Rhône-Poulenc-Verfahren wird als Katalysator ein mit Triphenylphosphantrisulfonat (TPPTS) komplexierter Rhodiumkomplex (Kuntz-Cornils-Katalysator) verwendet. Durch die Substitution des Triphenylphosphanliganden mit Sulfonatgruppen besitzt der metallorganische Komplex hydrophile Eigenschaften. Der Katalysator ist durch die neunfache Sulfonierung sehr gut in Wasser löslich (etwa 1 kg l−1), jedoch nicht in der entstehenden Produktphase. Das wasserlösliche Triarylphosphansulfonat wird im etwa 50-fachen Überschuss eingesetzt, wodurch das Auswaschen des Katalysators, das sogenannte „Leaching“, effektiv unterdrückt wird. Als Edukte werden Propen sowie Synthesegas, das aus Wasserstoff und Kohlenmonoxid im Verhältnis 1,1:1 besteht, eingesetzt. Es kann aus verschiedenen von Erdöl unabhängigen Rohstoffquellen erhalten werden. Als Produkt entsteht ein Gemisch aus n- und iso-Butanal im Verhältnis 96:4. Die Selektivität zum n-Aldehyd ist hoch, Nebenprodukte wie Alkohole, Ester und höhersiedende Fraktionen werden kaum gebildet.
Das Ruhrchemie/Rhône-Poulenc-Verfahren ist das erste kommerzialisierte Zwei-Phasen-System, in dem der Katalysator in wässriger Phase vorliegt. Im Fortgang der Reaktion bildet sich eine organische Produktphase aus, die mittels Phasenabscheidung kontinuierlich abgetrennt wird, wobei die wässrige Katalysatorphase im Reaktor verbleibt.
In diesem Verfahren werden in einem Rührkesselreaktor das Olefin und das Synthesegas von unten in den Reaktor geführt und die Phasen des Reaktionsgemisches intensiv durchmischt. Der entstehende Rohaldehyd wird am Kopf abgezogen. Im Phasenseparator wird die organische von der wässrigen Phase getrennt. Die wässrige katalysatorhaltige Lösung wird über einen Wärmetauscher vorgewärmt und wieder in den Reaktor gepumpt. In einem Stripper wird das überschüssige Olefin durch Synthesegas in Abwesenheit eines Katalysators von der organischen Phase getrennt und dem Reaktor wieder zugeführt. Die freiwerdende Reaktionswärme wird über Wärmetauscher zur Prozessdampferzeugung genutzt.
Der erzeugte Prozessdampf wird zur anschließenden Destillation der organischen Phase zur Trennung in iso- und n-Butanal genutzt. Der Destillationssumpf wird über einen Fallfilmverdampfer erwärmt und der Destillation wieder zugeführt. Potentielle Katalysatorgifte, die über das Synthesegas in die Reaktion eingeführt werden, werden mit dem Aldehyd abgetrennt. Dadurch kommt es zu keiner Anreicherung von Katalysatorgiften, die aufwändige Feinreinigung des Synthesegases kann daher entfallen.
In Oberhausen wurde 1984 eine Anlage gebaut, die im Jahr 1988 und nochmals 1998 auf eine Produktionskapazität von 500.000 t/Jahr Butanal erweitert wurde. Dabei werden 98 % des Propens umgesetzt und eine hohe Selektivität erzielt. Während des Prozesses geht weniger als 1 ppb Rhodium verloren.
BASF-Verfahren
Im Hydroformylierungsverfahren der BASF (BASF-Oxoverfahren) werden meist höhere Olefine eingesetzt. Als Katalysator dient Cobaltcarbonylhydrid. Der Katalysator wird von der flüssigen Produktphase durch Sauerstoff vom formal negativ geladenen Co−1 zum wasserlöslichen Co2+ oxidiert und durch Zugabe von wässriger Ameisen- oder Essigsäure abgetrennt. Dadurch bildet sich eine wässrige Phase aus, die das Katalysatormetall in Form seines Salzes enthält. Die wässrige Phase wird abgetrennt und das Cobalt in den Prozess zurückgeführt. Etwaige Verluste werden durch Zugabe frischer Cobaltsalze ausgeglichen. Eine Reaktion bei niedriger Temperatur führt zu einer erhöhten Selektivität zum linearen Produkt. Das Verfahren wird bei einem Druck von etwa 30 MPa und in einem Temperaturbereich von 150 bis 170 °C durchgeführt.
Exxon-Verfahren
Das Exxon-Verfahren oder Kuhlmann- oder PCUK-Oxoverfahren, dient zur Hydroformylierung von C6- bis C12-Olefinen. Zur Katalysatorrückgewinnung wird die organische Produktphase mit wässriger Natronlauge oder Natriumcarbonatlösung versetzt. Durch Extraktion mit Olefin und Neutralisation durch Zugabe von Schwefelsäurelösung unter Kohlenstoffmonoxiddruck wird das Metallcarbonylhydrid wiedergewonnen. Dieses wird mit Synthesegas ausgestrippt, vom Olefin aufgenommen und zum Reaktor zurückgeführt. Das Verfahren wird bei einem Druck von etwa 30 MPa und bei einer Temperatur von etwa 160 bis 180 °C in Gang gesetzt.
Shell-Verfahren
Das Shell-Verfahren nutzt Cobaltkomplexe mit Phosphanliganden zur Hydroformylierung von C7- bis C14-Olefinen. Die entstehenden Aldehyde werden direkt zum Fettalkohol weiterhydriert. Diese werden vom Katalysator über Kopf abdestilliert und der Katalysator wird als Sumpfprodukt erhalten und kann wieder in den Prozess zurückgeführt werden. Das Verfahren besitzt eine gute Selektivität zu linearen Produkten, die als Tensidrohstoffe Verwendung finden. Es wird bei einem Druck von etwa 4 bis 8 MPa und in einem Temperaturbereich von etwa 150 bis 190 °C durchgeführt.
UCC-Verfahren
Das UCC-Verfahren, auch als Low-Pressure-Oxo-Verfahren (LPO) bezeichnet, nutzt einen in hochsiedendem Dicköl gelösten Rhodiumkatalysator für die Hydroformylierung von Propen. Die Reaktionsmischung wird in einem Fallfilmverdampfer von flüchtigen Bestandteilen getrennt. Die flüssige Phase wird destilliert und n-Butanal über Kopf vom der Katalysatorphase entstammenden Dicköl getrennt. Bei diesem Verfahren wird ein Druck von etwa 1,8 MPa in einem Temperaturbereich von etwa 95–100 °C eingesetzt.
Literatur
- Arno Behr: Angewandte homogene Katalyse. Wiley-VCH, 2008, ISBN 978-3-527-31666-3.
- Maurizio Taddei, Andrè Mann (Hrsg.): Hydroformylation for Organic Synthesis. Springer, Heidelberg, New York, Dordrecht, London, 2013, ISBN 978-3-642-45059-4
- Boy Cornils: Hydroformylierung (Oxo-Synthese). In: J. Falbe, U. Hasserodt: Katalysatoren, Tenside und Mineralöladditive, Georg Thieme Verlag, 1978, ISBN 3-13-552601-1.
- Armin Börner, Robert Franke: Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, 2016, ISBN 978-3-527-33552-7


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 13.01. 2026