Nukleotidsequenz
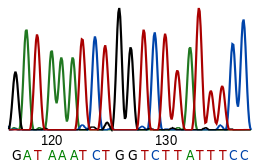
Die Nukleotidsequenz ist die Abfolge der Nukleotide einer Nukleinsäure, üblicherweise Desoxyribonukleinsäure (DNS, englisch DNA) oder Ribonukleinsäure (RNS, englisch RNA). Deren verschiedene Grundbausteine (Desoxyribonukleotide bzw. Ribonukleotide) sind gewöhnlich unverzweigt im Polymer verkettet und enthalten unterschiedliche Nukleinbasen. Die Sequenz seiner Nukleotide gibt die Primärstruktur eines Polynukleotides wieder und wird für DNA- oder RNA-Einzelstränge jeweils angegeben durch die Abfolge ihrer Nukleinbasen, die Basensequenz.
Bei der Notation werden für die Nukleinbasen der Nukleotide die Anfangsbuchstaben ihrer Bezeichnungen verwendet: für Adenin A, Guanin G, Thymin T, Uracil U und Cytosin C. Bei der DNA kommen die vier Basen Adenin, Guanin, Thymin und Cytosin vor, bei der RNA die vier Basen Adenin, Guanin, Uracil und Cytosin.
Übereinkunftsgemäß wird die Basensequenz vom 5′-Ende zum 3′-Ende des Stranges notiert, in der gleichen Richtung 5′→3′, in der die Polymerase die Nukleinsäure aus Nukleotiden synthetisiert.
Bestimmung
Die Nukleotidsequenz einer DNA wird durch DNA-Sequenzierung ermittelt. Basensequenzen von DNA werden unter anderem in großen öffentlichen Sequenzdatenbanken wie z.B. GenBank gespeichert. RNA wird nicht direkt sequenziert. Stattdessen wird sie mittels Reverser Transkriptase in eine DNA (cDNA) kopiert, die dann sequenziert wird.
Statistische Analyse
Dargestellt als Symbolfolge lassen sich Basensequenzen von DNA oder RNA gut untersuchen. Statistische Untersuchungen können beispielsweise die Häufigkeit sogenannter n-Tupel in der Sequenz vergleichen, also das Vorkommen von Teilfolgen der Länge n. So taucht im menschlichen Genom zum Beispiel die Folge CG bzw. das Tupel (C,G) insgesamt deutlich seltener als eines der anderen fünfzehn 2-Tupel auf (CG-Suppression). Lokale und regionale Häufigkeitsverteilungen verschiedener Nukleotidfolgen können auch erste Hinweise auf eventuelle Funktionen bestimmter DNA-Abschnitte geben. So wird bei CG beispielsweise nach Häufungen der CpG-Dinukleotide im DNA-Strang gesucht, den sogenannten CpG-Inseln, und deren Methylierungsmuster untersucht. In diesem Fall wird ein aus zwei Basen geformtes Sequenzmotiv gesucht.
Nach Drei-Basen-Sequenzmotiven wird gesucht, wenn mögliche Startcodons oder Stopcodons dargestellt werden sollen. Beispiele für etwas längere Sequenzen sind die möglichen Bindungsstellen für Ribosomen, wie die Shine-Dalgarno-Sequenz bei prokaryoten oder die Kozak-Sequenz bei eukaryoten Lebewesen. Bestimmte Nukleotidsequenzen spielen auch für den Terminator der Transkription eine wichtige Rolle, wie ebenso für den Startpunkt, an dem eine RNA-Polymerase mit der Transkription beginnt, beispielsweise in der Promotorregion eines Gens die TATA-Box.
Siehe auch


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 18.09. 2025